Immunocore PLC
Published: Aug 25, 2022 9:00 AM EST
Last Update: Aug 29, 2022 9:00 PM EST
Preface: Thanks to everyone who recently subscribed. This article took some time to write up but will remain free on blackseedbio.com. If you would like to support the website or show your appreciation, you can donate by clicking here. I am now also on Buy Me a Coffee and Patreon. If you would like to contact me about anything, please click here. Sharing the website with others also helps out a lot and I appreciate it very much. Thanks for your support.
TweetCONTENTS
Capital Structure History Management Board of Directors Ownership Product & Pipeline T Cell Receptors, T Cell Immunology & ImmTAX TCRs & MHC MHC gene diversity TCR technology history The ImmTAC/ImmTAV/ImmTAX platform Mechanism of Action Tebentafusp (KIMMTRAK) A little about Uveal Melanoma Tebentafusp Pivotal IMCgp100-202 study Commercialization KIMMTRAK approvals Uveal Melanoma market KIMMTRAK launch Uveal Melanoma global pipeline Tebentafusp for cutaneous melanoma IMC-C103C (RG6290) MAGE-A4 competition IMC-F106C PRAME competition IMC-I109V Hepatitis B virus and treatment challenges A functional cure for CHB Initial IMC-I109V data CHB global pipeline IMC-M113V A Background of HIV Reservoirs of HIV infection Some notes on other potential uses for TCR bispecifics PatentsCapital Structure
| IMCR ADS | in millions, $ (except Price) |
Last update |
|---|---|---|
| Price per ADS | 55.83 | Aug 25, 2022 |
| Shares outstanding | 48 | Q222 |
| Market cap | 2,657 | |
| Cash+Equivalents | 393 | Q222 |
| Debt | 0 | Q222 |
| Enterprise value | 2,264 |
History
Founding and Early History: Seed-stage
Immunocore was spun out of German biotech company, MediGene, in 2008. MediGene had acquired its T-cell receptor (TCR) platform from Avidex, a company that spun out of Oxford in 1999.
Avidex was developing TCR therapeutics and other immune modulating drugs at the time of its acquisition by MediGene. Suffice to say, MediGene was not after their TCR program. The former CEO of Avidex, James Noble, would become the founding CEO of Immunocore and pick up where Avidex left off.
Around the same time, James Noble and co. would also form sister company, Adaptimmune (ADAP), which today focuses on cellular TCR therapies. He would remain CEO of both companies until March 2014, when he would step down from his position at Immunocore and focus his efforts on Adaptimmune. He would continue to support Immunocore as a non-executive director.
Prior to his departure from Immunocore as an executive, Noble and the team would land collaboration deals with Big Pharma players like GSK (June 2013) and Genentech (June 2013).
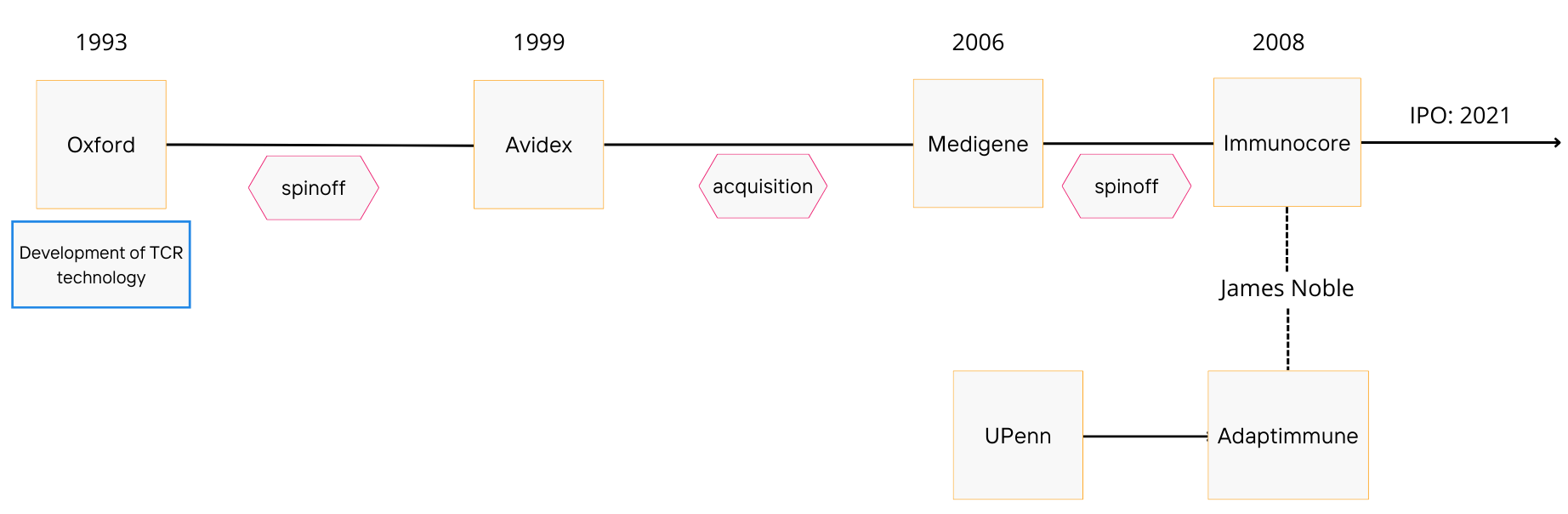
The work on TCRs at Oxford was pioneered by Bent Jakobsen, head of the Immune Receptor Group at the Institute of Molecular Medicine from 1993-2000. He is also a co-founder of Avidex, Immunocore and Adaptimmune, and has served as CSO at all three.
If it is not clear by now, the histories of Immunocore and Adaptimmune are inextricably intertwined. The two companies were also both initially funded by the likes of British hedge fund manager George Robinson, as well as Ian Laing, Nick Cross, and others. In 2013, the two companies would also share the same executive chairman, Dr Jonathan Knowles. Immunocore and Adaptimmune would begin to diverge in the mid-late 2010s, with Adaptimmune focusing on TCR cellular therapies, and Immunocore on soluble TCR products.
The Forster Era: Finding Water
In January 2015, Eliot Forster would succeed James Noble as CEO. Forster's time at the helm would be pivotal for Immunocore as they would begin to advance their lead candidate into Phase II and enter a new phase of growth.
In July that year, soon after Forster's entry, the company would raise $320 million in Series A financing. New investors included Woodford Investment Management, Malin Corporation, Eli Lilly and RTW Investments. There was some hype surrounding this as it was the largest financing round of a private life sciences company in Europe (at the time).
The company would also receive a $40 million investment from the Gates Foundation in September 2017. Eliot Forster would remain as CEO until February of 2018, leaving the seat to Andrew Hotchkiss in an interim role for about a year.
AstraZeneca Restructuring and the Bahija Jallal Era: Sprouting
In October 2018, a Type B (EOP) meeting with the FDA was held to discuss the registrational pathway for Immunocore's lead candidate, tebentafusp. Around the same time, restructuring at AstraZeneca and retiring of the MedImmune brand would open up new opportunities for the company.
In prime position, Immunocore would gain many new former AstraZeneca/Medimmune executives beginning early 2019. Former President of MedImmune Bahija Jallal would join the company as CEO, and Former SVP Head of AstraZeneca's IO franchise and Head of Early Stage Oncology at Medimmune, David Berman, would join as Head of R&D.
Koustubh Ranade, former VP of Translational Medicine at MedImmune and Mohammed Dar, former VP Clinical Development (Oncology) at MedImmune would also join Immunocore later in 2019, as Head of Translational Medicine and Head of Clinical Development & CMO, respectively.
Others following in from AstraZeneca would include Shaad Abdullah, former Director, Clinical Development at MedImmune, joining as Executive Director, Clinical Development in April 2019, and JoAnn Suzich, former Microbial Sciences Therapeutic Area Head at AstraZeneca, joining as Head of R&D in April 2020.
Once again, new financing would support the growing team. In March 2020, the company would secure $130 million in Series B financing. That round was led by General Atlantic, investing $75 million, with other new investors including CCB International, JDRF T1D Fund, Rock Springs Capital, Terra Magnum Capital Partners, and WuXi AppTec. Existing investors such as Eli Lilly, RTW Investments and the Bill & Melinda Gates Foundation would grow their positions. At this point Immunocore was valued at ~$640 million.
The company was incorporated in England and Wales in January 2021, and an IPO was completed on February 9, 2021, selling 11,426,280 ADSs at a price of $26 per ADS, each ADS representing one ordinary share. Gross proceeds were $297 million. An additional $15 million was raised concurrently in a private placement to the Gates Foundation.
Management
CEO: Bahija Jallal, since January 2019
Former President of MedImmune @ AstraZeneca, 2013-2019, Senior VP R&D at MedImmune 2009-2013. Prior to this, she was VP, Drug Evaluation & Translational Medicine at Chiron from 2005-2006 (until its acquisition by Novartis). She holds a PhD in Physiology and was a post-doctoral fellow at the Max Planck Institute of Biochemistry.
Head of R&D: David Berman, since January 2019
He was Senior VP, Head of Oncology at MedImmune from 2015-2017 and SVP, Head of AstraZeneca's IO Franchise from 2017-2018. Prior to that, he held multiple roles at Bristol Myers Squibb spanning 10 years from 2005-2015, with his last role as VP, Head of IO Exploratory Development from 2013-2015. He holds an MD PhD and completed his fellowship at Johns Hopkins in Pathology.
CCO/Head of Commercial: Ralph Torbay, since February 2021
He was Global Head of Hematology, Marketing at AstraZeneca from 2019-2021, Global IO (Imfinzi) Brand Lead for Lung Cancer from 2017-2019 and Global Launch Director (Tagrisso) from 2016-2017. Prior to that, he lead commercialisation of Arzerra in 1L CLL at Novartis (2015-2016) and worked on the launches of Farydak and Tasigna from 2014-2016.
Head of Clinical Development & CMO: Mohammed Dar, since April 2019
He was former VP, Clinical Development, Oncology, R&D at MedImmune from 2013-2019, leading clinical trials relating to durvalumab (anti-PD-L1) and moxetumomab (anti-CD22 immunotoxin). Prior to that, he spent 9 years at GSK, most recently as Senior Director, Cancer Research from 2011-2013. He holds an MD from Duke, and completed his Fellowship in Hematology and Oncology at the NCI.
Chief Technology Officer & Head of Pipeline and Platform Research: Annelise Vuidepot, since company founding
She has played a key role in the development of the ImmTAC platform that is central to Immunocore. She was a Senior Scientist at Avidex in from 2000-2003, and has been part of the senior leadership team since January 2017.
Head of Research: JoAnn Suzich, since April 2020
She was the former Microbial Sciences Therapeutic Area Head at AstraZeneca (from Jan 2019-July 2019), and had 31 years of experience prior to that at MedImmune. She holds a PhD in Biochemistry from Purdue.
CFO & Head of Strategy: Brian Di Donato, since May 2020
He was SVP and CFO at Achillion Pharmaceuticals from August 2018 until its acquisition by Alexion.
VP of Business Development: Stephen Megit, since company founding.
He has played a key role in delivering partnerships with Genentech, GSK, MedImmune an Eli Lilly. He holds a PhD in Experimental Medicine.
Board of Directors
Chairman: Professor Sir John Bell, appointed March 2015
He is the Regius Professor of Medicine at Oxford University and Chairman of the Office for the Strategic Coordination of Health Research. He was a founding director at Avidex. He currently serves on the boards of Roche and Genentech and previously had a role on the scientific advisory board at AstraZeneca. He also chairs the scientific committee of UK Biobank and the Global Health Scientific Advisory Board of the Bill and Melinda Gates Foundation. He is also one of the three Life Science Champions for the UK, reporting directly to the Prime Minister.
Travis Coy, appointed September 2019
Current VP, Head of Transactions and M&A, Corporate Business Development at Eli Lilly. He was responsible for leading and managing business development transactions for Lilly's cardiometabolic disease, drug delivery and medical devices.
Siddharth (Sid) Kaul, appointed June 2022.
He retired from Novartis as Group Treasurer and Head of Business Planning and Analysis in April 2021.
Kristine Peterson, appointed November 2017
She was most recently the CEO of Valeritas. Prior to that, she was Company Group Chair at Johnson & Johnson and has in total more than 30 years experience in the biopharmaceutical industry.
Professor Peter J. Ratcliffe, appointed October 2020
He is a physician scientist, currently the Director of Clinical Research at The Francis Crick Institute and Director for the Target Discovery Institute and Distinguished Scholar of the Ludwig Institute for Cancer Research at the University of Oxford. He was awarded the Nobel Prize for Physiology or Medicine in 2019 alongside William G Kaelin of Harvard and Gregg L Semenza of Johns Hopkins for their work in hypoxia.
Roy S. Herbst, appointed February 2021
He is a physician scientist, and was previously on Immunocore's SAB.
Rob Perez, appointed September 2019
He is an Operating Partner at General Atlantic since January 2019, playing a key role in Immunocore's Series B Financing.
CEO, Bahija Jallal, appointed January 2019
Ownership
As summarised below.
| Name | Shares beneficially owned | Market value ($, m) | % of SO | Reporting date |
|---|---|---|---|---|
| General Atlantic | 5,322,575 | 283 | 11% | July 20 2022 |
| Baker Bros | 4,685,691 | 250 | 10% | July 15 2022 |
| RTW Investments | 3,996,154 | 213 | 8% | June 30 2022 |
| Rock Springs Capital | 2,671,652 | 142 | 6% | June 30 2022 |
| Eli Lilly | 2,548,145 | 136 | 5% | Dec 31 2021 |
| Malin Life Sciences Holdings | 2,389,989 | 127 | 5% | Dec 31 2021 |
| Nicholas John Cross | 2,369,610 | 126 | 5% | Dec 31 2021 |
| Ian Laing | 2,358,650 | 126 | 5% | Dec 31 2021 |
| George Edward Silvanus Robinson | 2,153,520 | 115 | 5% | Dec 31 2021 |
| FMR | 1,360,384 | 72 | 3% | June 30 2022 |
| Avidity Partners | 1,350,000 | 72 | 3% | June 30 2022 |
| Wellington Management | 1,265,912 | 67 | 3% | June 30 2022 |
| Bill & Melinda Gates Foundation | 576,923 | 31 | 1% | June 30 2022 |
Product & Pipeline
As summarised below.
| Product | MOA | Indication |
|---|---|---|
| KIMMTRAK (tebentafusp) | gp100-targeted TCR-anti-CD3 scFv | Unresectable or metastatic UM in HLA-A*02:01-positive patients |
| Candidate | MOA | Proposed Indication | Phase |
|---|---|---|---|
| Tebentafusp | gp100-targeted TCR-anti-CD3 scFv | Advanced melanoma | II/III |
| IMC-C103C | MAGE-A4-targeted TCR-anti-CD3 scFv | NSCLC, gastric, HNSCC, ovarian, SS | I+ |
| IMC-F106C | PRAME-targeted TCR-anti-CD3 scFv | Breast, endometrial, ovarian, SCLC | I+ |
| IMC-I109V | HBV-Env-targeted TCR-anti-CD3 scFv | HBV cure | I |
| IMC-M113V | HIV-Gag-targeted TCR-anti-CD3 scFv | HIV cure | I |
T cell Receptors and T cell Immunology
TCRs & MHC
T cell receptors (TCR) are central to the activation, regulation and function of T cells. They are highly variable and made up of two chains (alpha and beta in 95% of T cells). They resemble the Fab (fragment) of immunoglobulins, with two variable and two constant domains. However, they are never secreted, and are in fact unstable in solution.
In addition, TCRs can only recognise short processed peptides bound to major histocompatibility complex (MHC) molecules, unlike immunoglobulins that recognise antigens in their native form. T cells also do not undergo somatic hypermutation, so TCRs do not become 'affinity maturated' like immunoglobulins. However, TCRs have a greater overall level of diversity than immunoglobulins (up to 1018 possible antigen-specific TCRs compared to 1014 possible antibody specificities are generated.
T cell development from heamatopoietic stem cells occurs in the thymus with stringent selection before release into the periphery (98% of thymocytes dying before release).
The repertoire of mature T cells are sculpted after various TCR component rearrangements in intrathymic progenitor cells. Self-reactive T cells (that bind self-antigen:MHC molecules too tightly) are eliminated (eliminating auto-reactive T cell receptors). At the same time, T cells which do not recognise peptide bound to self-MHC sufficiently do not receive the survival signal needed to survive thymic development. This process produces T cells that have an intrinsic affinity to self MHC molecules, but at a binding strength insufficient for activation.
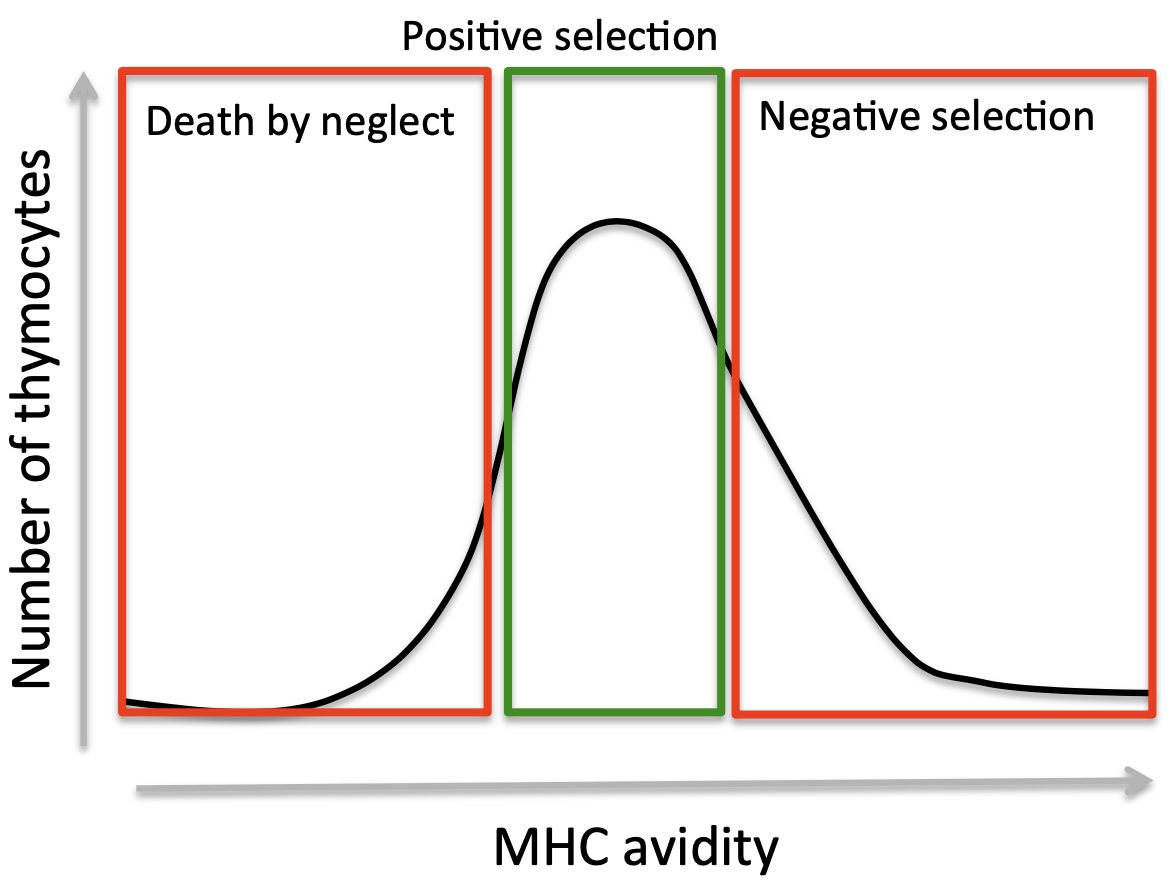
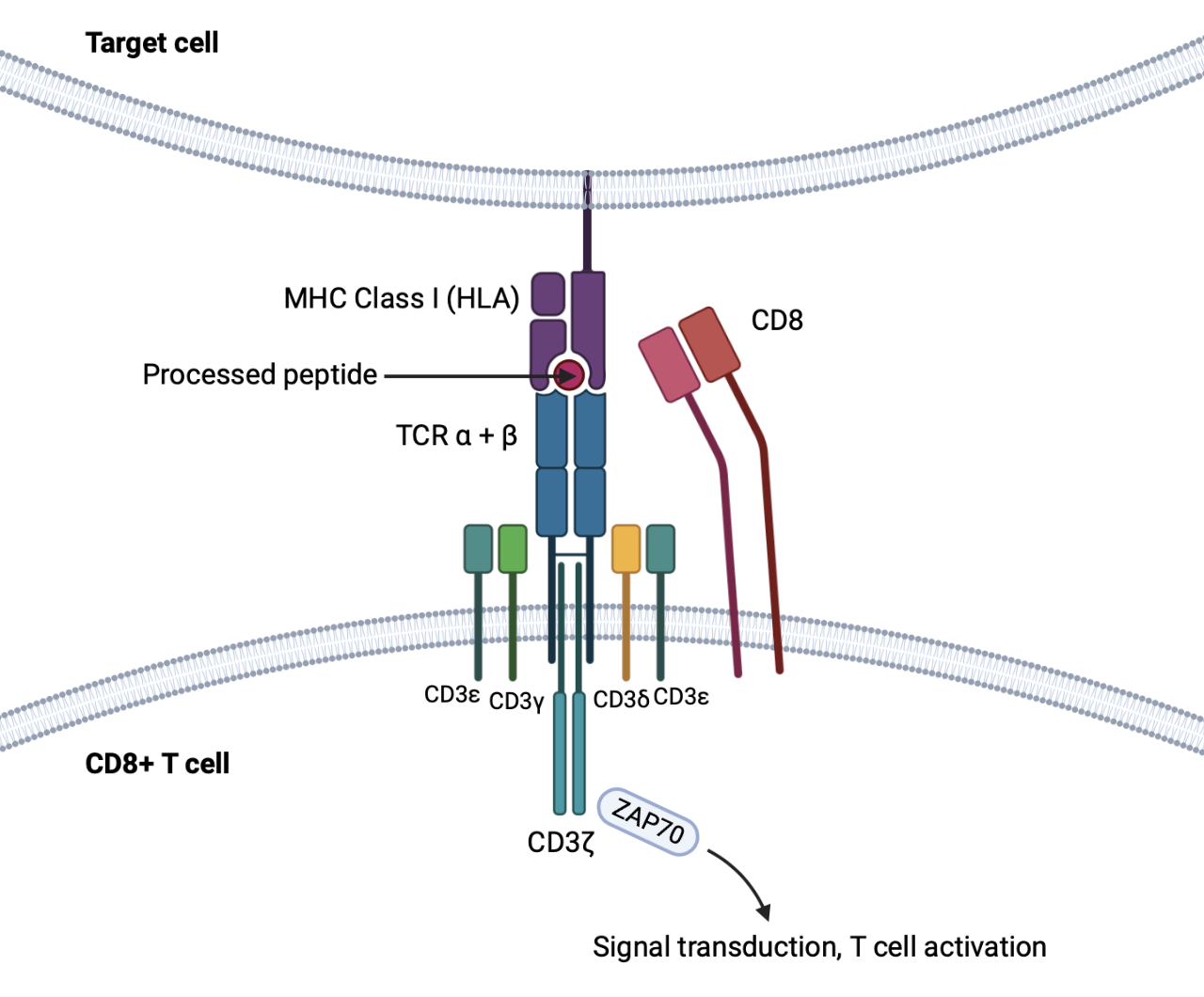
MHC molecules (or HLA in humans) come in two types, MHC Class I (expressed on almost all nucleated cells and recognised by CD8+ T cells) and MHC Class II (expressed on professional antigen presenting cells such as dendritric cells, macrophages, and recognised by CD4+ T cells).
MHC molecules bind short peptides (8-11 amino acids long) from proteins that have been processed by cellular protesomes.
Antigen presentation occurs constitutively and informs about the 'health' of the cell. In the normal state, self-antigens are displayed on the surface, but this does not activate T cells due to insufficient binding (as a result of the tolerance built during T cell development).
Alterations in the proteome (i.e by viral infection or cell transformation) are reflected on MHC-I molecules and can be sensed by CD8+ T cells (through TCR engagement). CD8+ T cells are cytolytic, and upon stable binding to peptide-MHC-I (pMHC), release molecules such as perforin and granzyme B to induce apoptosis in the infected/transformed cell.
Upon TCR-pMHC engagement, conformational changes in the TCR result in the formation of TCR-CD3 microclusters and so called "immunological synapse". The intracellular tails of CD3 molecules contain immunoreceptor tyrosine-based activation motif (ITAM), sequences that become phosphorylated to activate tyrosine kinases and trigger a signalling cascade that leads to cellular activation.
MHC Gene Diversity
MHC genes (HLA alleles in humans) display high genetic polymorphism, with over 100 different forms (alleles) of certain HLA genes. This polymorphism affects the peptide binding domains and can alter or abolish binding of certain peptides, as well as certain TCRs. Importantly, TCRs are not only specific for peptides, but also for MHC molecules.
Each T cell receptor recognises a peptide and only one of the MHC molecules within an individual. Note that a single individual can express multiple HLA class I and class II alleles, but the T cells are always restricted by self-HLA products.
Of the various HLA types, HLA-B has the highest degree of polymorphism, with over 3000 variants reported.1 The HLA-A2 type is the most prevalent, with expression in ~50% of Caucasians, ~47% of Hispanics, ~35% of African-Americans and ~36% of Asian/Pacific Islander.2
From the HLA-A2 type, the 01 subtype (HLA-A*2:01) is the most common and found in 96% of Caucasians, 73% of Hispanic, 59% of African-American and 53% of Asian/Pacific Islander. These numbers are important as the TCRs that Immunocore develops, like native TCRs, can only recognise specific HLA types. This limits the number of people eligible for treatment.
TCR Technology History
The TCR technology (soluble, recombinant TCRs) at the center of Immunocore was developed at Oxford University in the 1990s. This led to the formation of Avidex, later acquired by MediGene.
A patent filed in 1999 (WO/1999/060120) (Bent Jakobsen of Oxford is one of the inventors) makes claims to soluble TCRs with mention of potential therapeutic utility. An additional patent filed in 2003 (US7569664) makes claims to specific linker sequences that enable disulfide bond formation between the TCR alpha and beta segments.
A paper published by Jakobsen's group in Protein Engineering that year, entitled "Stable, soluble T-cell receptor molecules for crystallization and therapeutics" reports the production of these soluble TCRs, targeted to the HLA-A*02:01-NY-ESO peptide complex.
The first patent exploring soluble TCRs is US6080840, filed 1995, a continuation of a patent filed in 1992. One of the inventors on that patent is Alfred E. Slanetz, former President & CEO of Bluebird Bio, who was at that time completing his PhD on T cell receptors at Yale. He shares inventorship with Alfred Bothwell, and their paper "Heterodimeric, disulfide-linked alpha/beta T cell receptors in solution" was published in 1991 in the European Journal of Immunology. They built off the work of a Howard Hughes/Stanford group led by Mark M Davis.
A Harvard group lead by Jack L. Strominger, (who collaborated with the group at Oxford) also worked on TCRs. They published a paper titled "Binding of soluble natural ligands to a soluble human T-cell receptor fragment produced in Escherichia coli" in 1994. A patent of Strominger (WO1996018105) describes the single chain TCR and the E. coli expression system used to produce it. Immunocore are also using an E. coli expression system to produce their TCR bispecifics.
Another company, Sunol Molecular Corporation, was also working on producing soluble TCRs at the time (patent WO1999018129). They received a $93,000 grant from the NIH in 2001. That company was spunoff into Altor Bioscience, which continued to work on TCRs (patent US20180086810) but nothing significant came out of this work. Altor was acquired by ImmunityBio in 2017 (which later merged with NantKwest in 2021).
The ImmTAC/ImmTAV/ImmTAX Platform
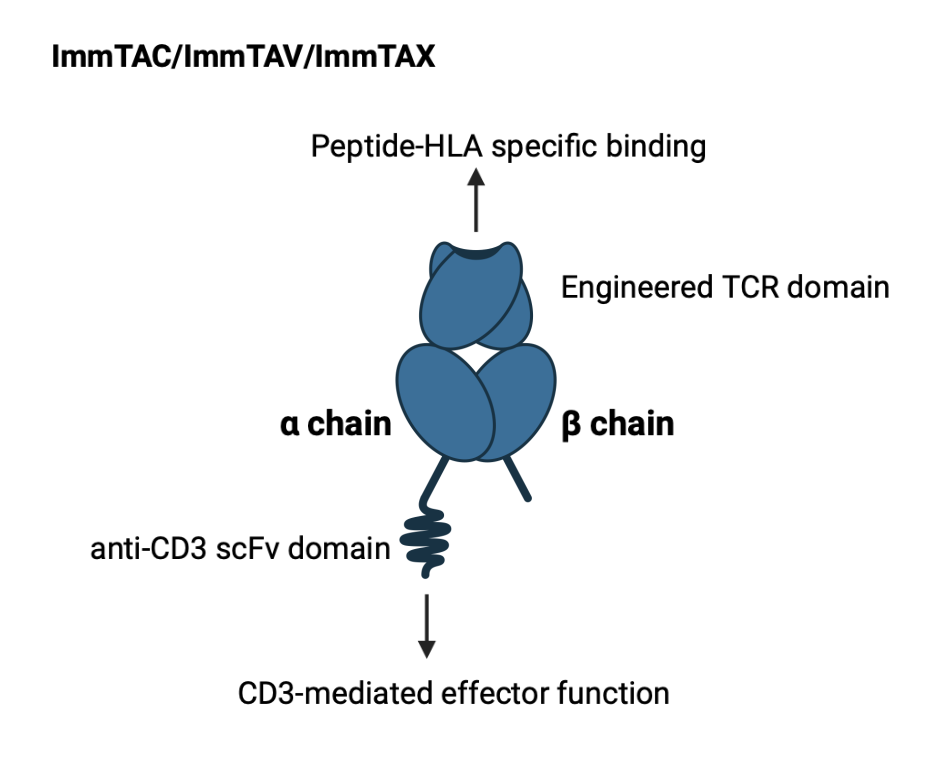
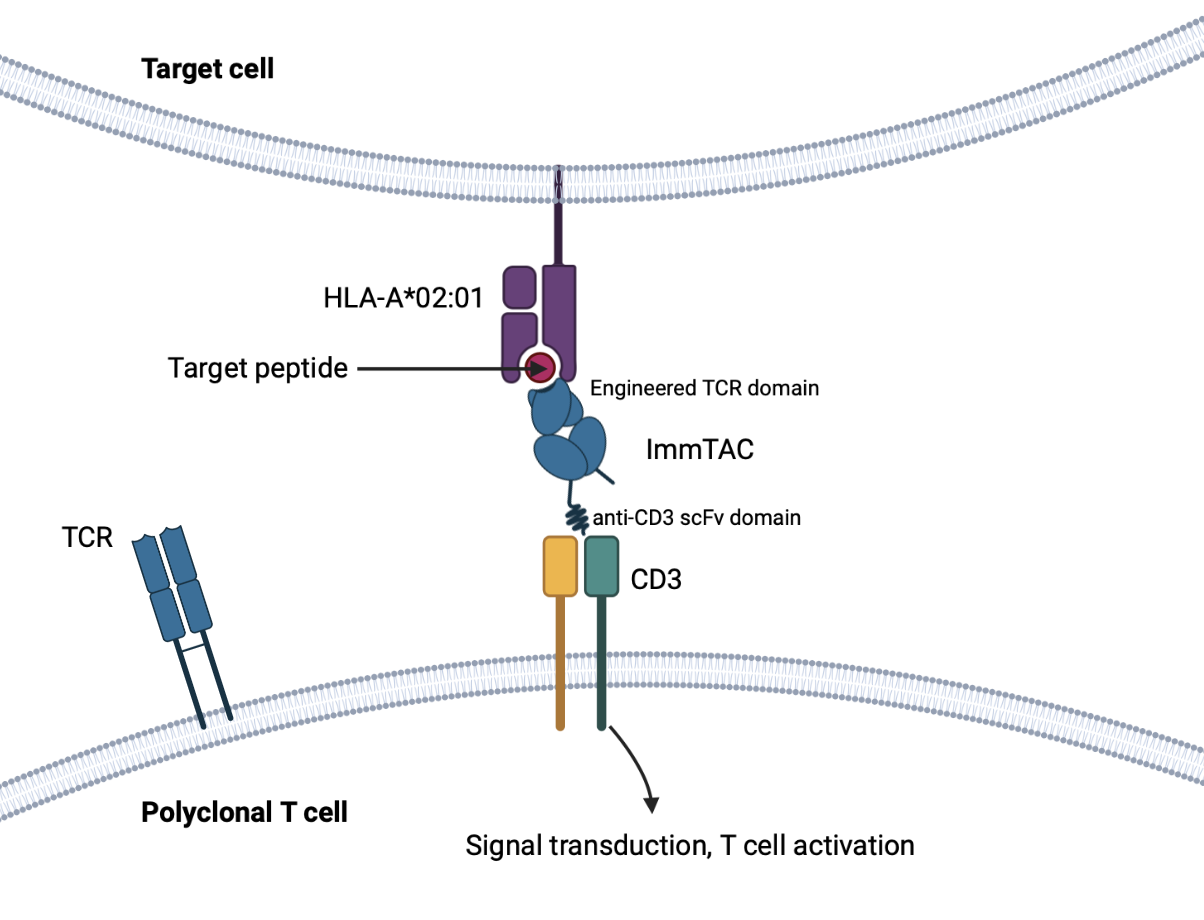
Immune mobilizing monoclonal TCRs against cancer (ImmTACs) or against viruses (ImmTAVs) or X disease (ImmTAX) are the names Immunocore gives to their high affinity bispecific TCR/anti-CD3 scFv products. These products are the culmination of decades of research and incorporate various aspects of TCR immunology. These aspects are well summarized in a paper titled "Engineering soluble T-cell receptors for therapy3".
The TCR domain is stabilized by a non-native disulfide bond linking its two constant regions. The variable loops can bind their target with affinity in the picomolar range. The scFv domain binds CD3 with lower affinity (in the nanomolar range, 1000-fold weaker), to ensure that activity is driven by affinity to the target pHLA and minimize off-target effects.
These ImmTAX theoretically have wider application than monoclonal antibodies, bispecifics or CAR-T cells as they are able to target transformed or viral intracellular proteins (after processing and display on HLA molecules). This aspect is valuable in diseases where native TCRs are insufficient and/or for a variety of reasons, not generating the immune responses they should be.
ImmTAX molecules may also be vastly superior to native TCRs in contexts where peptide presentation is reduced (such as due to the effects of a viral protein), or if a molecular switch has reduced affinity to MHC (such as in cancer)4.
Additionally, these soluble TCRs have technical advantages over adoptive T cell therapy, which are typically autologous (derived from the patient), technically demanding, and more expensive to produce.
Mechanism of Action
The TCR domain binds to infected or transformed cells expressing or displaying specific target antigens (through pHLA). The anti-CD3 scFv domain induces effector responses at this site that leads to polyclonal T cell activation (which do not need to have native TCRs specific to the target pMHC).
This leads to a broad immune response and T-cell mediated cytotoxicity against the infected or transformed cell.
Think like Amgen's BITEs (bispecific T cell engagers), but for HLA-presented antigens.
The key paper to read here is titled "Monoclonal TCR-redirected tumor cell killing" published in Nature Medicine in 2012, where the team at Immunocore describe various engineered TCRs targeting peptides of gp100, MAGE-A3, MART-1 and NY-ESO-1, bound to HLA-A*0201 (or HLA-A*0101 in the case of MAGE-A3 peptide), each with ability to potently activate CD8+ T cells and induce lysis of tumor cells.
Tebentafusp (KIMMTRAK)
A little about uveal melanoma
Uveal melanoma (UM) represents 85-90% of ocular melanomas. Patients are initially treated with curative intent by surgery and radiation, but 50% of patients develop metastatic disease, usually metastatizing to the liver (80-90% of cases).
UM presents with a different genetic profile than cutaneous melanoma. Unlike cutaneous melanoma, where driving mutations are commonly found at the BRAF and NRAS loci, the most common driver mutations in UM are activating mutations of GNAQ (42.2% of cases) and GNA11 (32.6%), as well as inactivating mutations in BAP1 (31.5%, 84% of metastasizing tumors). BRAF/NRAS mutations are mostly absent/rare in UM, so treatments that target this pathway are ineffective.5
There have been no proven therapies that improve survival in mUM until KIMMTRAK. UM is highly resistant to systemic chemotherapy. Nonetheless, in the unresectable/metastatic setting, patients are typically treated with therapies approved for cutaneous melanoma, such as ipilimumab (CTLA-4), pembrolizumab (PD-1) or an anti-CTLA-4/PD-1 combination, despite showing limited efficacy. However, the approval of KIMMTRAK is changing the landscape.
Tebentafusp
Tebentafusp is an ImmTAC with a high affinity gp100280-288-targeting domain (Kd=24 pM, in vitro) fused to an anti-CD3 scFv effector domain (Kd = 38 nM, in vitro). Its target, gp100 (glycoprotein 100) is a melanocyte differentiation antigen and highly expressed in melanocytic cells.
The gp100 peptide that tebentafusp binds with high affinity are residues 280-288, a peptide that has been shown to have particularly high affinity to subtype HLA-A*02:01. Its restriction to this HLA subtype makes it an attractive target for TCR therapy. Note that it is not the only epitope that is presented on HLA molecules and recognised by T cells, but 280-288 is a well-characterized one. Other epitopes of gp100 CTLS have been shown to recognise include 154-162 and 457-466.6
Tebentafusp has a molecular weight of 77 kDa and is produced efficiently by recombinant DNA in E. coli cells in facilities located in Denmark (drug substance, AGC) and Germany (finished product, Baxter Oncology).
It was approved by the FDA on January 26, 2022 for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma. This was the first recombinant TCR therapeutic approved for treatment in the world. It was also the first therapy approved for unresectable or metastatic uveal melanoma, and the first treatment shown to extend overall survival in this disease in a randomized trial.
FDA approval was primarily based on a data set of 378 newly-diagnosed patients (in the first line), comparing tebantefasup with investigator's choice (either single-agent pembrolizumab, single-agent ipilimumab or dacarbazine; 82% of which were administered pembrolizumab/Keytruda).
This trial restricted treatment in the control arm to these three options. In consequence, ipilimumab/nivolumab combination therapy was precluded, a treatment which has grown in popularity over the last few years in mUM due to better efficacy (although there have been no head to head clinical trials to prove this definitively).
An important observation in this trial was that the survival benefit experienced in patients treated with KIMMTRAK was independent of whether cells had low or high expression of gp100, indicating high potency (and a lack of need for high numbers of target pHLA copies for activity). This is a more significant finding for the platform in general in terms of its applicability to other diseases, as expression of gp100 is typically high at all stages in UM.7
Tefanbasup is administered once-weekly by intravenous infusion with a prime-dosing regimen to mitigate CRS (a common AE of CD3-mediated immune activation). The first two doses (day 1 and day 8) are 20 mcg and 30 mcg respectively, before escalation to 68 mcg on day 15.
In the first three doses, patients are to be monitored for at least 16 hours post-infusion for CRS, clinically significant hypotension and hypoxia. If the patient does not experience Grade 2 or worse hypotension, subsequent doses are to be performed in appropriate outpatient settings with 30 minutes of post-infusion monitoring.
Pivotal IMCgp100-202 study, a randomized, open-label trial
The trial (NCT03070392) enrolled 378 newly-diagnosed uveal melanoma patients confirmed positive for HLA-A*2:01 positive. The primary outcome measure was OS, and patients were recruited from 56 sites spanning 14 countries.
Safety
The most common treatment-related adverse event was CRS. In the study, 89% of patients treated with KIMMTRAK (n=245) experienced CRS (all grades). However, the rate of grade 3+ CRS was low (0.8%) and typically manageable. Permanent discontinuation due to CRS occured in only 1.2% of patients.
Other toxicities were mainly rash (83%, 18% Gr3+), pyrexia (76%, 4% Gr3+), pruritis (69%, 5% Gr3+), fatigue (51%, 6% Gr3+), nausea (49%, 2% Gr3+) and chills (47%).
Most AEs were manageable and decreased following the dose escalation stage. In a post-hoc analysis, 74% of patients treated with tebentafusp did not require corticosteroids and only 10% received them on a single day.
Efficacy
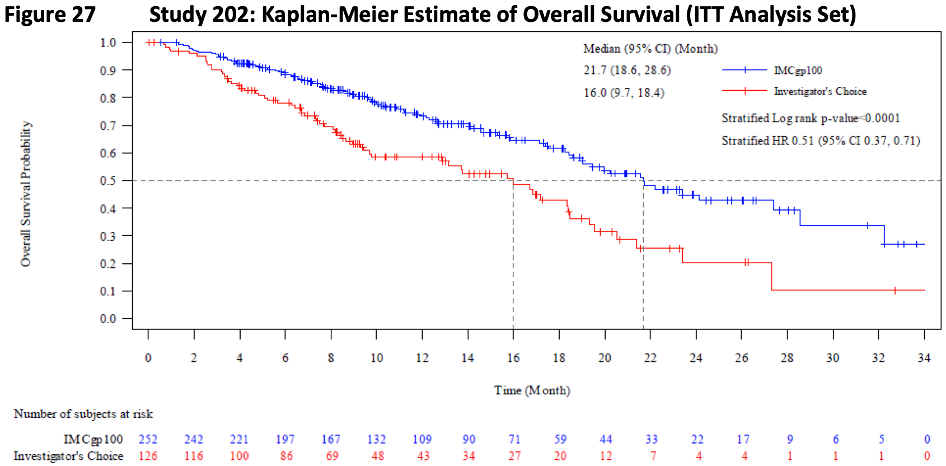
The median PFS was 3.3 months (vs 2.9 months investigator's choice, HR=0.73, p=0.0139). The overall response rate (ORR) as measured by RECIST was also not clinically meaningful (9% vs 5% control). However, the key here is that while tumor response rates by RECIST were marginal, patients that did respond had significantly improved survival (median OS 21.7 months vs 16 months control, HR=0.51; 95% CI: 0.37, 0.71; p<0.0001).
The hazard ratio (HR) in those with progressive disease as best response was even more accentuated at 0.43 (OS 15.3 months vs 6.5 control). These data provided evidence of significant clinical benefit beyond RECIST and were foundational for regulatory approval.
Miscellaneous
Of note, 33% and 29% of patients receiving tebantafusp in study 102 (the Phase I/II study) and study 202 respectively developed anti-drug antibodies (ADAs), with a median onset time to ADA formation 6-9 weeks after initial treatment. The neutralisation capacity of these ADAs are unknown, but clearance of tebantafusp increased with higher titer ADAs. While the onset time seems relatively close to the PFS (3.3 months), exploratory analyses (with limited data) suggested no clinical significance (i.e no observed effect of decreased overall survival).
Nonetheless, Immunocore was issued a post-marketing commitment to assess the presence of neutralizing ADAs of all patient samples that tested positive for binding antibodies in these studies and to evaluate their clinical impact on PK, safety and efficacy. That evaluation report was submitted in February 2022.
Commercialization
KIMMTRAK Approvals
Uveal Melanoma Market
According to curemelanoma.org, there are 2,500 new cases of uveal melanoma in the U.S annually. SEER Program data (Surveillance, Epidemiology, and End Results) collected by the National Cancer Institute (NCI) from 1973-2013 suggests an incidence of 5.2 per million and has remained stable over the time period. This would translate to about ~1,700 new cases a year in the U.S.
The incidence rate is similar in Europe, Canada and Australia, which would translate to about ~1,000 cases in Western Europe, ~350 cases in the UK, 200 cases in Canada and 150 cases in Australia every year. In total, there are about 3,400 new cases collectively in these populations per year.
According to SEER, 98% of uveal melanoma cases are diagnosed in Caucasians. KIMMTRAK is indicated for HLA-A*02:01 positive patients, which is about 48% of Caucasians. This would mean about ~1,600 of the total number of cases each year would be eligible for treatment (800 in US, 500 in Western Europe, 170 in UK, 100 in Canada, 70 in Australia).
Immunocore estimates 1,000 metastatic UM patients per annum in the US and Western Europe who test positive for HLA-A*02:01.
KIMMTRAK Launch
Immunocore signed an exclusive multi-regional agreement with Medison Pharma on October 18 2021, for the commercialization of KIMMTRAK in Canada, 20 markets across Central Eastern Europe and Israel. This deal was extended on May 23 2022 to include Australia and New Zealand. The drug was approved in Canada June 8 2022 and in Australia on June 3 2022. The details of this agreement have not been released.
KIMMTRAK was launched immediately in the US after FDA approval on Jan 26 2022. KIMMTRAK is priced at $18,760 per vial. Each vial is single-dose and patients are treated once a week. The median duration of exposure to drug in study 202 was 5.3 months (0.3-33 months), or ~23 doses. Assuming the duration of treatment is equivalent in the real world (and patients do not return to treatment with KIMMTRAK), the average cost of treatment per patient in the U.S. comes to ~$430,000, with max revenue at $215mm (assuming 500 eligible metastatic UM patients).
On April 1 2022, marketing authorization was given by the European Commission. The first patient infused with commercial drug occured less than a week after price listing. All 50+ patients from the early access program (EAP) transitioned to commercial supply in May. Approval in the UK was given on June 8 2022. EU/UK pricing is confidential, however a draft appraisal by NICE (The National Institute for Health and Care Excellence, UK) published in July 2022 recommended against treatment due to uncertain cost-effectiveness (although this is not NICE's final guidance and may change after consultation).
KIMMTRAK generated $10.1 million and $29.2 million in net product revenues in the first and second quarters, respectively. In the second quarter, $7.1 million came from Europe, most of which came from Germany (2 months). Commercial drug supply came into France towards the end of the quarter.
| in millions, $ | Q122 | Q222 |
| KIMMTRAK US sales | 10.1 | 22.1 |
| KIMMTRAK EU sales | - | 7.1 |
| Global KIMMTRAK sales | 10.1 | 29.2 |
In the last quarterly conference call, it was reported that 40% of patients being treated with KIMMTRAK today are in the first line. CCO Ralph Torbay also stated the company's goal of reaching over 1,000 patients by 2025. This would translate to about $215 mm in annual revenues from the U.S. (assuming 500 eligible metastatic UM patients/year treated at a price of $430,000 per patient), and $135 mm in annual revenues in the ex-U.S. market (assuming 500 patients/year at an average price of $270,000 per patient).
In April 2022, KIMMTRAK was added to the US NCCN (National Comprehensive Cancer Network) guidelines as a Category 1 treatment for metastatic uveal melanoma.
In June 2022, KIMMTRAK was added to the ASCO Rapid Recommendations Updates to replace ASCO Guidelines for the treatment of HLA-A*02:01-positive patients with metastatic uveal melanoma.
Recommendation 4.1.1 reads: Previously untreated HLA-A*02:01-positive patients with metastatic uveal melanoma should be offered tebentafusp (Type: Evidence-based, benefits outweigh harms; Evidence quality: Moderate; Strength of recommendation: Strong).
Recommendation 4.1.2 reads: For all patients with uveal melanoma other than those addressed by Recommendation 4.1.1, no recommendation for or against any specific systemic therapy may be made at this time. Patients should be offered or referred for enrollment in clinical trials where possible (Type: No recommendation; Evidence quality: Low; Strength of recommendation: Not applicable).
Uveal Melanoma Global Pipeline
N.B. A comprehensive table is now available in the Data tab
Foghorn Therapeutics has a BRG/BRM1 inhibitor, FHD-286, in Phase I for mUM (NCT04879017). That mechanism of action is through GNAQ/GNA11. A report on Foghorn published a few months ago on Blackseed Bio can be read here.
IDEAYA Biosciences has a PKC inhibitor, darovasertib (IDE196), in a Phase II clinical trial for GNAQ/11+ mUM in combination with crizotinib. PKC is functions downstream of GNAQ/11 A clinical update from this program is expected in September 2022.
Another PKC inhibitor, Array Biopharma/Pfizer's sotrastaurin (AEB071) was in Phase I for mUM in combination with a MEK inhibitor in the mid 2010s (NCT01801358), but that trial was terminated "for scientific reasons."
Vanquish Oncology in partnership with Genentech have an ongoing Phase Ib/II trial (NCT04589832) testing a Procaspase 3 activator, PAC-1, in combination with TKI, entrectinib (Rozlytrek).
Roche has an anti-TYRP1/CD3 T cell engager, RO7293583 in Phase I for unresectable TYRP1+ melanomas, including UM. That trial (NCT04551352) is ongoing.
Linnaeus Therapeutics has a G protein-coupled estrogen receptor (GPER) agonist, LNS8801, in Phase I/II for the treatment of mUM as monotherapy or in combination with pembrolizumab. That trial (NCT04130516) began in late 2019.
Verastem Oncology is testing their RAF/MEK clamp in combination with a FAK inhibitor in GNAQ/GNA11-mutated mUM. FAK was identified as a synthetic lethal gene candidate with GNAQ activation. That trial is investigator-sponsored and in Phase II (NCT04720417).
Bellicum had an HLA-A2 restricted PRAME-directed TCR expressing autologous T cell product, BPX-701 in trial for previously treated AML/MDS or UM. Two mUM patients were enrolled, one of which experienced a PR. That program was discontinued, but the results can be read here.
BioMed Valley Discoveries' ERK inhibitor, Ulixertinib, was in Phase II for mUM, but that trial did not demonstrate efficacy. ASCO abstract here.
Tebentafusp for Cutaneous Melanoma
Gp100 is expressed on tumors of melonocytic origin, including cutaneous melanomas (CM). A phase Ib/II trial of tebentafusp in previously-treated metastatic CM patients is ongoing and recruiting patients (NCT02535078). Patients must be HLA-A*02:01+ to be enrolled.
There are 5 arms in this trial: single-agent tebentafusp, tebentafusp in combination with durvalumab (PD-L1 inhibitor) or tremelimumab (CTLA-4 inhibitor), or as triplet therapy in combination with the two. An additional arm exploring tebentafusp by subcutaneous injection was also included.
The most recent data from this trial was published in June at ASCO 2022 from 112 patients (Abstract can be accessed here).
97/112 (86%) were relapsed or refractory to prior anti-PD1 (80% also received ipilimumab).
33 of 97 patients in any arm who received durvalumab >= 10 mg/kg had a 1-yr OS of 79% and a 2-yr OS of 34%. The median OS was 20 months. 64/97 patients who received a durvalumab dose <10 mg/kg had a 1-yr OS of 53%, a 2-yr OS of 24% and a median OS of 13 months. OS in the triplet arm (teme+durva+treme) was similar to the teme+durva double therapy. The benchmark 1 year approximate survival is 55%, so this data is encouraging, but will need further trial.
Immunocore is planning to initiate a Phase II/III trial in previously treated advanced CM in Q4 of this year. Patients will be treated with tebentafusp as a single agent or in combination with an anti-PD-1.
IMC-C103C (RG6290)
This is a MAGE-A4:HLA-A*02:01-targeting ImmTAC, currently in Phase I for the treatment of HLA-A*02:01-positive patients with advanced tumors expressing MAGE-A4. The drug is being developed in collaboration in a co-development/co-promotion deal with Genentech/Roche.
MAGE-A4 is expressed in multiple solid tumors, such as esophageal, ovarian cancer, NSCLC and HNSCC. Immuncore estimates >75,000 total HLA-A*02:01 positive patients with metastatic MAGE-A4+ tumors annually who could potentially benefit, mostly with NSCLC, ovarian and HSNCC.
Results from the dose escalation portion showed an expected toxicity profile, 62% experiencing chills, 57% pyrexia and 52% CRS (in the 90-240 mcg dose range with pharmacodynamic activity, n=21). 33% at this dose experienced Grade 3/4 neutropenia. No treatment-related AEs led to discontinuation.
Strong T-cell activity was observed at doses >=90 mcg, with IL-2, IL-6, IFNgamma increases, and induction observed mainly in MAGE-A4 positive tumors (indicating appropriate targeting).
There were three confirmed PRs (out of 11 evaluable MAGE-A4-positive tumors), one with HNSCC with a duration of response (DOR) of 2+ months (ongoing) and two with ovarian cancer with a DOR of 8.3 months and 4.4+ months (ongoing), respectively. Two other patients (both with ovarian cancer) had tumor shrinkages, with target lesion reductions of 44% and 81% respectively (new lesions formed however).
Most of the patients at the data cut-off in the Phase I trial have had low or no MAGE-A4 expression.
The first expansion arm in high grade serous ovarian has been initiated with a dose set at 140 mcgs. Updated Phase I clinical data are expected in Q4 of 2022.
Immunocore's presentation of the data can be accessed here.
MAGE-A4 Competition
Immatics is developing a soluble bispecific TCR, IMA401, targeting an HLA-A*02-presented peptide derived from both MAGE-A4 and MAGE-A8. This drug is being developed in collaboration with BMS and entered Phase I in May 2022.
Roche has an HLA-A2-MAGE-A4 x CD3 bispecific T cell engager, RG6129 in Phase I for the treatment of HLA-A*02:01 positive patients with MAGE-A4+ solid tumors (NCT05129280).
These two drugs likely represent the most direct competition to Immunocore's program.
Other competition include adoptive T cell therapies targeted to MAGE-A4, such as Adaptimmune's ADP-A2M4, Immatics' CAR-T, IMA201, and Zelluna Immunotherapy's allogeneic TCR based NK cell therapy.
IMC-F106C
A PRAME-targeting ImmTAC for HLA-A*02:01-positive patients. PRAME is expressed in various solid tumors such as NSCLC, SCLC, TNBC, endometrial, ovarian, cutaneous and uveal melanoma. It is a negative prognostic marker. There are potentially 150,000 patients who could be eligible for treatment (i.e HLA-A*02:01 with PRAME-expressing tumors)
Initial Phase I data from 20+ patients treated at active doses are being presented at ESMO in September as an oral presentation.
PRAME Competition
The most direct competition is Immatics' soluble TCR bispecific, IMA402, also targeted to HLA-A*02-bound-PRAME peptide. The candidate is in preclinical development with planned Phase I initiation in 2023.
IMC-I109V
IMC-I109V is an ImmTAV designed to eliminate Hepatitis B virus (HBV)-infected hepatocytes. It engages HBV ENV-derived-peptide bound to HLA*A2:01.
Hepatitis B Virus and Treatment Challenges
Hepatitis B virus (HBV) is a DNA virus with features that are somewhat similar to retroviruses (like HIV). Its chronicity is the result of integration into the host genome and/or existence as extrachromosomal covalently closed circular (ccc) DNA.
The HBV cccDNA is a nucleic replication intermediate that can continue to produce progeny virus. Stably integrated HBV DNA is not replicative, but can continue to produce viral RNAs and proteins. Both cccDNA and integrated DNA are sources of persistent HBsAg production and impairment of HBV-specific T cell responses (although cccDNA is the main source of the problem).
While many vaccines and antivirals to HBV exist, there is no functional cure for those who are chronically infected (i.e achieving seroclearance of HBsAg). Long term antiviral therapy typically has little effect on HBsAg levels, and is also problematic for patient compliance.
The WHO estimates 300 million people in the world are living with chronic Hepatitis B (CHB), mostly residing in Africa, Asia, and South America. Up to 2.4 million and 13.3 million cases of CHB are in the US and Europe respectively.
The AASLD recommends indefinite antiviral therapy for adults with HBeAg-negative immune-active CHB (i.e persistent HBV infection that is uncleared by the immune response). Without appropriate management, 15-25% of people with CHB will develop advanced liver disease and/or hepatocellular carcinoma (HCC). In 2019, CHB resulted in 820,000 deaths, mostly from these sequelae.
A Functional Cure for CHB
There are three main barriers to a functional cure in CHB:
1. Reservoir of cccDNA and/or integrated DNA (in hepatocytes) that is uncleared by antiviral therapy
2. Dysfunctional T cell responses (immune exhaustion) driven by chronic antigen exposure with limited killing of infected hepatocytes
3. Insufficient B cell responses
Immunocore is developing IMC-I109V to redirect non-exhausted polyclonal T cells to HBV-displaying cells. If successful, IMC-I109V could eliminate the reservoir of HBV in infected hepatocytes and act as a functional cure.
Initial IMC-I109V Data
Initial data were presented at the 2022 EASL International Liver Congress, from three patients treated at a starting single dose of 0.8 mcg. Surprisingly, such a low dose resulted in a transient decrease in HBsAg in two of three patients with concomitant increases in ALT (by 11-15% during days 3-15 post infusion). Elevations in ALT are indicative of hepatocytic death and are an indication of pharmacodynamic activity. These levels returned to baseline within 3 weeks.
Lymphocyte margination (i.e trafficking into the tissue) was also evident in one of the patients (47% reduction in lymphocyte counts 24 h post infusion), consistent with the mechanism of action. This patient (pt #3) also had an almost 6-fold increase from baseline in serum IL-6. IL-6 generally promotes T cell proliferation, differentiation and an inflammatory response.8 It is pleiotropic, also having direct effects on hepatocytes as a mitogen (promoting liver regeneration) and in the production of acute phase proteins (like CRP).9,10 The duration of the acute phase response is normally 24-48 hours.
Note this is a very complicated area of immunobiology, with multiple feedback loops and location-specific effects, and more data are needed to build up strong evidence for activity. Clearance of HBsAg will be key here.
The trial was initiated with a low starting dose, as a conservative dosing schedule will be necessary to reduce the rate of hepatocytic lysis and limit potentially serious liver damage-related AEs. Based on preclinical work, 0.8 mcg was determined to be the MABEL (minimum anticipated biological effect level). This is a subtherapeutic dose, so it was surprising to see any efficacy at this dose, even partial. This is preliminary evidence for the drug's potency. It was also positive to see that there were no signs of CRS at this dose (although this will likely be a bigger factor at higher doses).
A poster presentation of the initial data can be viewed here.
An important point to note is that if successful, IMC-I109V will only be available for HLA*A2:01 positive patients. However, while HLA*A2:01 is less prevalent in non-Caucasian populations, it is still the most common. There is nonetheless a great number of people infected with CHB around the world, with no curative or functionally curative therapies available.
CHB Global Pipeline
There are a number of companies developing drugs for CHB. Below are some of the treatment approaches companies are taking. Note this list is not exhaustive.
Therapeutic Vaccines
GSK has an HBV viral vector vaccine + protein adjuvant with GSK3528869A aimed at restoring HBV-specific T cell responses in CHB. A Phase I/II trial (NCT03866187) initiated in Q1 2019 and is ongoing.
Altimmune has HepTcell - nine synthetic HBV-derived peptides formulated with a TLR9-based adjuvant designed to reverse immune tolerance/exhaustion in a similar MOA to Immunocore's ImmTAX. Their candidate is in a Phase II trial with a data readout anticipated in H2 2023.
VBI Vaccines has a recombinant protein-based vaccine being developed in collaboration with Brii Biosciences, VBI-2601 being investigated in a Phase II trial with initial data expected in H1 2023. The drug is also being used as a combination therapy with VIR-2218, with data expected in H2 2022.
Immune Activators
Gilead is developing a TLR-8 agonist, selgantolimod (GS-9688) that has completed Phase II. An ongoing Phase II trial (NCT04891770) testing the drug in combination therapies (such as with nivolumab or Vir Biotechnology's siRNA candidate, VIR-2218) is currently ongoing.
Roche has a TLR-7 agonist, RG7854, in Phase I.
There are also a few checkpoint inhibitors in clinical trials to activate exhausted HBV-specific T cells. Ascletis Pharma, Gilead and Roche are each testing anti-PD-L1s as functional cures for CHB.
Oligonucleotides
GSK also has an antisense oligonucleotide (ASO) in-licensed from Ionis, bepirovirsen (GSK3228836), with Phase II data presented at EASL 2022. A Phase III trial is anticipated to begin in the first half of 2023. More on this can be accessed here. and here.
Replicor is also using oligonucleotides to inhibit the production of HBsAg. Their candidates, REP 2139 and REP 2165 are in Phase II.
Aligos Therapeutics has an siRNA, ALG-125755 with a Phase I trial initiating in Q4 2022.
Gene Editing
This treatment approach is in early development and can come with the typical risks associated with endonucleases/gene editing.
Excision Bio has a CRISPR/Cas9 system with a candidate, EBT107, in early preclinical development.
Precision BioSciences also has an in vivo gene editing program to knockout cccDNA and inactivate integrated HBV DNA. Their candidate is a nuclease delivered as mRNA in LNP, and their preclinical work was published in May 2022 in the paper "Targeting the hepatitis B cccDNA with a sequence-specific ARCUS nuclease to eliminate hepatitis B virus in vivo".
Miscellaneous
Aligos Therapeutics has a Class-I capsid assembly modulator, ALG-000184 in Phase I.
IMC-M113V
IMC-M113V is an ImmTAV directed to an HIV-Gag-peptide presented on HLA*A2:01 (specifically the p17 epitope, SLYNTVATL a.k.a. SL9). The goal of treatment here is to achieve a functional cure for HIV (i.e eliminate HIV reservoirs in latently infected cells).
HIV causes chronic infection by integration into the host genome and remaining in a latent state. There are 38 million people in the world infected with HIV with no cure available.
The current practice is to treat patients indefinitely with anti-retroviral therapy (ART) with the goal of sustaining low viral loads. Discontinuation of ART eventually leads to viral rebound due to persistent latent HIV reservoirs.
Achieving a functional cure will allow patients to discontinue ART permanently. While the ideal treatment would allow permanent discontinuation of ART, extended periods of time off ART may also be beneficial and may be required if small unreachable HIV reservoirs persist in the body.
A Background of HIV
The HIV genome is composed of three main structural genes: gag, encoding structural proteins of the capsid, matrix, core and nucleocapsid; pol, encoding viral enzymes (protease, reverse transcriptase, RNase H and integrase); and env, encoding envelope glycoproteins.
A high degree of variability exists for gag and env proteins, contributing greatly to immune escape. This is one of the issues that has limited the success of vaccines and may potentially limit the effectiveness of IMC-M113V, although the Gag peptide/epitope IMC-M113V targets is a highly conserved and functionally constrained (i.e mutation would result in dysfunction).
HIV enters cells through Env binding to CD4 and/or CCR5 and/or CXCR4 chemokine receptors. Receptor tropism is in part determined by HIV's large genetic diversity. The majority of cells infected are CD4+ T cells, but other non-CD4 expressing cells can also be affected (such as macrophages and dendritic cells) contributing to reservoir virus.
Reservoirs of HIV Infection
The size of the reservoir is largely determined by the time antiretroviral therapy (ART) is initiated, with earlier therapy significantly reducing reservoir size.11 HIV reservoirs can be found in:
1. Resting CD4+ memory T cells in the lymph nodes and blood: - integration into host DNA can occur in resting or terminally differentiated cells. These cells undergo homeostatic proliferation. HIV latency means viral protein expression may be limited.
2. Lymph node germinal centres as immune complexes trapped on follicular dendritic cells in B-cell follicles
3. Macrophages, dendritic cells, astrocytes, in tissue and lymph nodes
4. Anatomical reservoirs that are immune privileged such as the brain (microglia and astrocytes) and testes. These may contribute to residual viral replication
These are some of the potential difficulties in achieving a functional cure. If functional, IMC-M113V may not be able to target latent cells with inactive or very low HIV-1 viral protein expression (specifically Gag expression). The therapy may need to be combined with "shock" therapies that activate HIV expression.
There may also be dificulty in reaching immune-privileged sites where sanctuaries exist, although the capacity of these reservoirs to give rise to replication-competent virus has not been fully determined. Expression of MHC-I in these compartments may also be limited or absent without stimulation.
Immunocore's HIV program is funded by the Bill and Melinda Gates Foundation. As part of the agreement, Immunocore is required to make any successfully approved products from this program available at reduced prices in certain developing countries. Immunocore retains full development and commercial rights in non-developing countries.
Preclinical data can be read here.
The first patient in the trial was dosed on July 11 2022.
Some Notes on Other Potential Uses for TCR Bispecifics
Besides transformed and viral proteins, TCRs are theoretically capable of targeting any proteins from the cell proteome. This opens up a wide range of possible targets.
TCR therapeutics may have synergistic effects with MHC upregulators. Proteins that upregulate or increase the activity of proteosomal degraders in turn upregulate MHC-I expression. The Hepatitis C virus (HCV) core protein has been shown to do this.12
TCR therapeutics could also be combined with NK cell-targeted therapies. Cells that upregulate MHC-I can avoid NK cell detection (MHC-I is a ligand for inhibitory receptors on NK cells).13 In this context, the TCR bispecific can eliminate cells that have escaped detection by NK cells (with higher potency).
The anti-CD3 scFv domain can be exchanged for one that dampens the immune response. In this way, auto-antigen-specific (self-reactive) T cells can be inactivated or immunosuppressed. The TCR domain in this case would also serve as a decoy molecule (reducing the number of available pHLA for self-reactive TCR binding). Such a hypothetical drug could be used chronically for relief in autoimmune diseases.
Patents
As summarised below.
| Asset | Patent type | Earliest Expiry | Jurisdiction |
|---|---|---|---|
| Tebentafusp (KIMMTRAK) | COM, Dosing, Formulation | 2030, 2037, 2040 | All major markets |
| IMC-C103C | COM | 2037 | All major markets |
| IMC-F106C | COM | 2038 | All major markets |
| IMC-I109V | COM | 2040 | All major markets |
| IMC-M113V | - | - | - |
Disclosure: I am a shareholder of Immunocore as of the time of the publishing of this article. I may exit from this position at any given time without notice. Data presented in this article have been obtained from third-party publications and sources. I do not guarantee the accuracy of this data and all information presented here should be checked and verified accordingly. Readers of this article should each make their own evaluation and judgement of the mentioned companies and of the relevance and adequacy of the information provided. Readers should make other such investigation as deemed necessary. The article is intended for informational purposes. Investors and potential investors are requested to do additional research before investing in any of the companies mentioned in this article. All investments have risks of loss associated with them. Investing is very risky, highly speculative, and should not be done by anyone who cannot afford to lose the entire value of investment and without prior due diligence. Investors should evaluate the risks associated with each individual company before investing.
Medical Advice Disclaimer: The material presented on this website is not medical advice and not intended to be a substitute for professional medical advice, diagnosis or treatment. Always seek the advice of your doctor or other qualified health provider regarding a medical condition and never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Additional disclosure: This article contains trademarks, trade names, copyrights and data of Immunocore and other companies, which are the property of their respective owners.
Tweet
References
1. Robinson J., Halliwell J.A., Hayhurst J.D. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43(Database issue):D423–D431
2. Ellis, J. M., Henson, V., Slack, R., Ng, J., Hartzman, R. J., & Katovich Hurley, C. (2000). Frequencies of HLA-A2 alleles in five U.S. population groups. Human Immunology, 61(3), 334–340. doi:10.1016/s0198-8859(99)00155-x
3. Robinson, R. A., McMurran, C., McCully, M. L., & Cole, D. K. (2021). Engineering soluble T‐cell receptors for therapy. The FEBS Journal.doi:10.1111/febs.15780
4. Bianchi V, Bulek A, Fuller A, Lloyd A, Attaf M, Rizkallah PJ, Dolton G, Sewell AK, Cole DK. A Molecular Switch Abrogates Glycoprotein 100 (gp100) T-cell Receptor (TCR) Targeting of a Human Melanoma Antigen. J Biol Chem. 2016 Apr 22;291(17):8951-9. doi: 10.1074/jbc.M115.707414. Epub 2016 Feb 25. PMID: 26917722; PMCID: PMC4861463.
5. Chattopadhyay, C., Kim, D. W., Gombos, D. S., Oba, J., Qin, Y., Williams, M. D., … Patel, S. P. (2016). Uveal melanoma: From diagnosis to treatment and the science in between. Cancer, 122(15), 2299–2312.doi:10.1002/cncr.29727
6. Barker, A. B. H., Schreurs, M. W. J., Tafazzul, G., De Boer, A. J., Kawakami, Y., Adema, G. J., & Figdor, C. G. (1995). Identification of a novel peptide derived from the melanocyte-specific gp100 antigen as the dominant epitope recognized by an HLA-A2.1-restricted anti-melanoma CTL line. International Journal of Cancer, 62(1), 97–102.doi:10.1002/ijc.2910620118
7. Wagner, S. N., Wagner, C., Schultewolter, T., & Goos, M. (1997). Analysis of Pmel17/gp100 expression in primary human tissue specimens: implications for melanoma immuno- and gene-therapy. Cancer Immunology, Immunotherapy, 44(4), 239–247.doi:10.1007/s002620050379
8. Korn, T., & Hiltensperger, M. (2021). Role of IL-6 in the commitment of T cell subsets. Cytokine, 146, 155654.doi:10.1016/j.cyto.2021.155654
9. Schmidt-Arras, D., & Rose-John, S. (2016). IL-6 pathway in the liver: From physiopathology to therapy. Journal of Hepatology, 64(6), 1403–1415. doi:10.1016/j.jhep.2016.02.004
10. Bode, J. G., Albrecht, U., Häussinger, D., Heinrich, P. C., & Schaper, F. (2012). Hepatic acute phase proteins – Regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. European Journal of Cell Biology, 91(6-7), 496–505. doi:10.1016/j.ejcb.2011.09.008
11. Massanella, M., Bender Ignacio, R. A., Lama, J. R., Pagliuzza, A., Dasgupta, S., Alfaro, R., … Chomont, N. (2021). Long-term effects of early antiretroviral initiation on HIV reservoir markers: a longitudinal analysis of the MERLIN clinical study. The Lancet Microbe, 2(5), e198–e209.doi:10.1016/s2666-5247(21)00010-0
12. Herzer K, Falk CS, Encke J, Eichhorst ST, Ulsenheimer A, Seliger B, Krammer PH. Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J Virol. 2003 Aug;77(15):8299-309. doi: 10.1128/jvi.77.15.8299-8309.2003. PMID: 12857899; PMCID: PMC165225.
13. Paul, S., & Lal, G. (2017). The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Frontiers in Immunology, 8. doi:10.3389/fimmu.2017.01124
Have feedback? Tweet or DM me @blackseedbio on X, or contact me here.
If you liked this article and want to get notified when updates are available, consider subscribing (it's free, and you can unsubscribe at any time).
Click here to donate.