Prelude Therapeutics
Last updated: Aug 15, 2021 1:00 PM EST
First posted: Aug 9, 2021 9:00 AM EST
CONTENTS
Background Management Ownership Pipeline PRT543 PRMT5 PRMT5 inhibitors GSK3326595 JNJ-64619178 PF 0639999 LLY-283 JBI-778 TNG908 MRTX9768 GSK3235025/EPZ0156666 Closely related competition PRT543 & Data Prelude's position PRT811 PRT811 Opportunity PRT1419 Intrinsic Pathway of Apoptosis Bcl-2 family of proteins Pro-survival proteins Venclexta & Beyond Bcl-2 Targeting MCL-1 Safety & The Cardiac Issue MCL-1 inhibitors AMG 176 AZD5991 MIK665/S64315 AMG 397 ABBV467 PRT1419 & Data Closely-related competition PRT2527 CDK9 inhibition CDK9 competition PRT2527 & Data PRT-SCA2 SMARCA2 & Synthetic Lethality SMARCA2 degraders Patents Finance & Upcoming Milestones ReferencesBackground
Prelude Therapeutics was founded in 2016 by Kris Vaddi, PhD, formerly a Founding Scientist, VP, and most recently, Senior Advisor at Incyte (2002-2016). From its inception in 2016, Kris has built a strong science-driven internal discovery team, with three ongoing clinical programs with in-house developed drugs, and three other pre-clinical programs with IND filings anticipated over the next year. The company is based in Wilmington, Delaware with 51 full-time employees (as of June 30 2021).
In the early days, the company was largely financed by Baker Bros and OrbiMed, purchasing Series A convertible preferred stock in two rounds in October 2017 and August 2018 for proceeds of $30 million. Prelude member of the Board and former CEO of Incyte, Dr Paul A. Friedman also invested.
Series B financing closed in May 2019 and March 2020, with all three of the above increasing their stake to raise an additional $60 million for the company.
In August 2020, the company raised an additional $50 million with a round of Series C financing, this time with Fidelity purchasing in.
An IPO was completed on September 25, 2020, pricing 8.3 million shares at $19, raising net proceeds of $158 million with an initial market cap at $807 million.
Management
The management team is closely tied to Incyte, with much of its C-suite previously holding positions at Incyte.
As above, CEO Kris Vaddi was a founding scientist at Incyte, initiating and leading the JAK research programs that led to the development and approvals of Jakafi (ruxolitinib) and Olumiant (baricitinib). Prior to founding Prelude Therapeutics, Kris founded and served as the CEO of Orsenix (2014-2016), a privately-held biotech that recently sold its clinical candidate, a form of arsenic trioxide to Syros Pharmaceuticals.
CSO Peggy Scherle, PhD, joined Prelude in April 2018. Like Kris, she was a founding scientist at Incyte that contributed to the development of drugs like Jakafi and Olumiant. She most recently held the position of Group Vice President, Discovery Biology and Preclinical Pharmacology at Incyte (2017-2018). Her prior roles at Incyte were Vice President, Preclinical Pharmacology (2014-2017) and Executive Director, In Vitro Biology (2011-2014). Before joining Incyte, Peggy held positions at DuPont Pharmaceuticals and Bristol Myers Squibb. She holds a PhD in Immunology from the University of Pennsylvania with postdoctoral training at the NIH.
EVP and Head of Chemistry, Andrew Combs, PhD, joined Prelude in April 2019. He also held several roles at Incyte from 2003-2019, most recently as Vice President of Discovery Chemistry (2015-2019). He first joined Incyte as Senior Director (2003-2006), and was an Executive Director from 2006 to 2015. Like Peggy, Andrew also held positions at DuPont and BMS prior to joining Incyte. He has a PhD in Organic Chemistry from UCLA and was a Howard Hughes Medical Institute post-doctoral fellow at Harvard University.
CMO Dr David Mauro, MD PhD, joined Prelude in May 2019. He was previously the CMO of Checkmate Pharmaceuticals (2016-2019), CMO of Advaxis (2014-2016), Executive Director at Merck (2007-2014) and Director at BMS. He contributed to the NDAs of Erbitux (cetuximab), Sprycel (dasatinib) and Sylatron (peginterferon alfa-2b). He received his MD PhD from Temple University.
EVP and Chief of Clinical Affairs, Dr Deborah Morosini, MD, MSW, joined Prelude in 2020. She was previously VP of Clinical Affairs and Patient Engagement at Loxo Oncology playing a key role in the launch of Vitrakvi (larotrectinib) and launch preparations for Retevmo (selpercatinib). She previously held positions at Foundation Medicine, AstraZeneca, Ardais.
EVP and Chief of Business Operations, Christopher Pierce, MBA, joined Prelude in 2020. He was previously VP and Head of Commercial at Loxo Oncology. He led the launch of Vitrakvi in partnership with Bayer and Retevmo. Prior to joining Loxo, Chris held roles at Pfizer (2007-2017), most recently as Senior Director and Global Commercial Lead of the Hematologic Malignancies Portfolio launching and managing numerous kinase inhibitors such as Xalkori (crizotinib), Sutent (sunitinib), Inlyta (axitinib), Bosulif (bosutinib) as well as ADCs such as Mylotarg (gemtuzumab ozagamicin) and Besponsa (inotuzumab ozogamicin). He worked at Millennium Pharmaceuticals from 2004 to 2006, managing market analytics for Velcade (bortezomib).
SVP and Head of Strategic Planning & Operations, Aimme Crombie, PhD, joined Prelude as VP, R&D Operations (2018-2020). Prior to this, she had numerous roles at Trevena, most recently as VP of Research, contributing to the development of numerous drugs for the treatment of CNS disorders. She was a scientist at Pfizer and Wyeth, and holds a PhD in Organic Chemistry from MIT.
VP, Early Development and Regulatory Affairs, Adam Shilling, PhD, joined Prelude in April 2019. Prior to joining Prelude, Adam held numerous roles at Incyte from 2007-2019, most recently as Executive Director of Global Regulatory Affairs where he led the GVHD franchise and JAK1 programs. He led the NDA submission and approval of Jakafi in acute GVHD and contributed to numerous other NDAs for Jakafi and Olumiant.
Board of Directors
Dr Paul A Friedman, since July 2016. He is the CEO of Madrigal Pharmaceuticals, former CEO of Incyte (2001-2014), President of R&D at DuPont-Merck (1994-1998), President of Dupont Pharmaceuticals Research Laboratories (1998-2001), and Senior VP at Merck Research Laboratories (1991-1994). Prior to this, he was an Associate Professor of Medicine and Pharmacology at Harvard Medical School.
Kris Vadi, CEO of Prelude.
Dr David Bonita, since July 2016. He is a General Partner at OrbiMed, and holds an MD/MBA from Columbia University.
Dr Victor Sandor, since May 2020. He was CMO of Array BioPharma (2014-2020), Senior VP for Global Clinical Development at Incyte (2014-2019), VP and CMO for Oncology at Biogen (2010-2014), and VP Global Oncology Drug Development at Incyte (2010-2014)
Mardi C. Dier, since August 2020. She was EVP and CFO/CBO of Portola Pharmaceuticals until its acquisition by Alexion.
Julian C. Baker, since January 2021. He is a Managing Member of Baker Brothers Investments.
Martin Babler, since July 2021. He was the President/CEO of Principia Biopharma from 2011 until its acquisition by Sanofi in October 2020, President/CEO of Talima Therapeutics (2007-2011), and held positions at Genentech (1998-2007), most notably VP Immunology Sales and Marketing.
Ownership
As summarised below.
| Name | Shares owned | Value (m) | % of total |
|---|---|---|---|
| Baker Bros | 10,123,824 | 326 | 21.6% |
| OrbiMed Advisors | 10,039,691 | 323 | 21.4% |
| FMR (Fidelity) | 6,252,107 | 201 | 13.3% |
| Kris Vaddi, CEO | 2,937,783 | 95 | 6.3% |
| State Street Corp | 873,376 | 28 | 1.9% |
| Vanguard | 867,420 | 28 | 1.8% |
| BlackRock | 681,725 | 22 | 1.5% |
| UBS Asset Management | 642,979 | 21 | 1.4% |
| HHLR Advisors | 626,502 | 20 | 1.3% |
| Price T Rowe Associates | 422,991 | 14 | 0.9% |
| Rock Springs Capital Management | 378,195 | 12 | 0.8% |
| Federated Hermes | 336,100 | 11 | 0.7% |
| TOTAL OF LISTED | 34,182,693 | 1,100 | 73% |
Pipeline
As summarised below. All drugs are wholly-owned.
| Candidate | Mechanism of Action | Indication | Phase |
|---|---|---|---|
| PRT543 | PRMT5 inhibitor | Selected solid tumors (ACC, HRD+), Selected myeloid malignancies (MF, MDS) | I |
| PRT811 | Brain-penetrant PRMT5 inhibitor | Glioblastoma, primary CNS lymphoma, CNS metastases | I |
| PRT1419 | MCL-1 inhibitor | AML, MDS, MM, NHL (Oral), Solid tumors (IV) | I |
| PRT2527 | CDK9 inhibitor | Solid tumors, Hematological malignancies | IND |
| PRT-SCA2 | SMARCA2 degrader | SMARCA4-mutant cancers | Preclin |
| PRT-K4 | Kinase | Solid tumors | Disc |
PRT543
PRT543 is the company's lead candidate currently in the expansion phase of an ongoing phase I trial (NCT03886831). The drug is a protein arginine methyltransferase 5 (PRMT5) inhibitor, designed at Prelude and chosen from a library of 600 compounds.
PRMT5
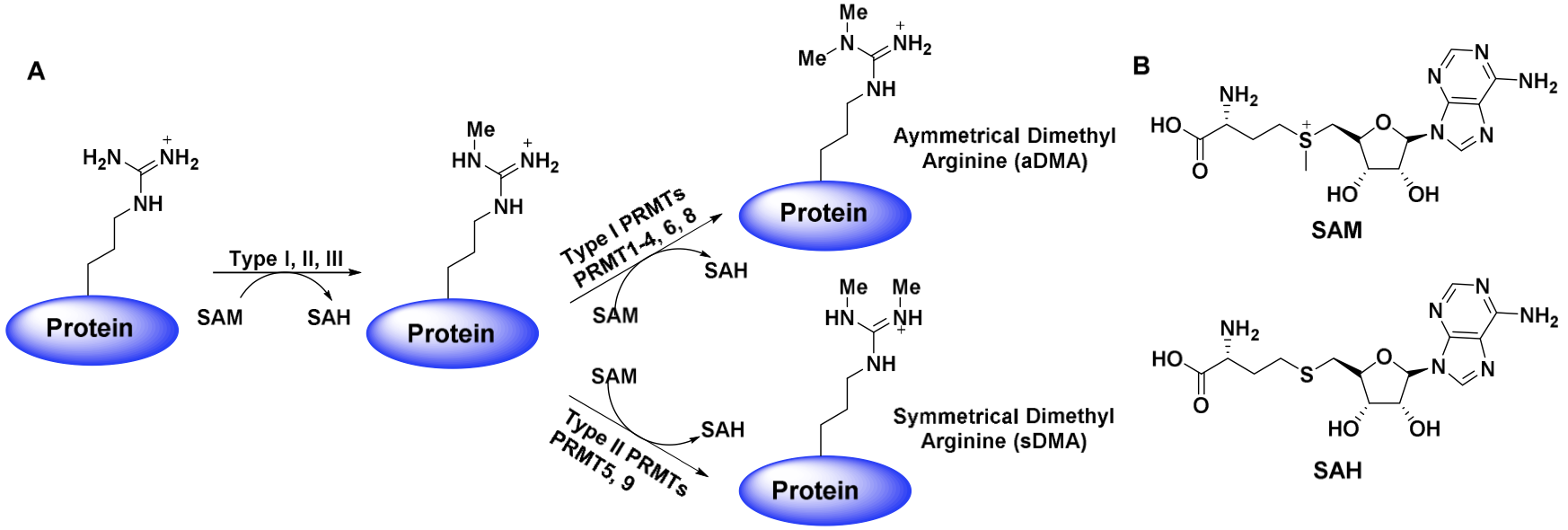
Short: PRMT5 is an enzyme that catalyzes the transfer of methyl groups to the arginine residues of various target proteins, such as histones, p53, E2F-1, NF-kB, and components of the spliceosome complex. Specifically, it symmetrically di-methylate the two terminal ω-guanidino nitrogens of arginine. Its target proteins include histones, p53, E2F-1, NF-kB and components of the spliceosome complex.
There are 9 members in the PRMT family, (PRMT1-9), that can be subdivided into Type I, II or III. All types of PRMTs catalyze the transfer of methyl groups from the co-factor, S-adenosylmethionine (SAM), or AdoMet, to the guanidinium nitrogen atoms of arginine residues. This process converts SAM to S-adenosyl-L-homocysteine (SAH). The difference however, between Type I and Type II PRMTs, is that Type II PRMTs (5, 9) symmetrically dimethylate their targets (forming symmetrically dimethylated arginine ((SDMA)) residues), whereas Type I PRMTs (1-4, 6, 8) asymmetrically dimethylate. Note that the Type III PRMT (PRMT7) is much less common and only catalyzes monomethylation.
PRMT5 in particular is differentiated from the other PRMTs in that it has a TIM Barrel domain preceding its SAM-dependent methyltransferase1. It is this domain that allows PRMT5 to form a stable complex with MEP50 (p44)2. permitting recruitment of a histone substrate peptide to PRMT5 (through the beta-barrel domain groove), for dimethylation and transcriptional corepression.
Of the Type II PRMTs (5 and 9), PRMT5 is thought to be the primary SDMA forming enzyme, with its deletion resulting in complete loss of SDMA in mouse embryo fibroblasts (MEFs). On the other hand, PRMT9 is a minor contributor to to the pool of SDMA, with its substrates likely limited and fewer in number.3
Data also suggest that PRMT5 has a role in methylation of pICln complex to generate SDMA in Sm proteins (the proteins that form the core of the spliceosomal complex with snRNAs for post-transcriptional modifications)4,5. This process regulates the efficiency of the assembly of snRNPs that make up the spliceosome, with a wide range of downstream effects.
PRMT5 has also been shown to co-localise with RAF proteins and modulate their catalytic activity through arginine methylation, limiting ERK1/2 signalling in the RAS-ERK signal transduction pathway6. It can also limit activity of BRG1 (SMARCA4) and BRM (SMARCA2), frequently dysregulated7,8 and key components of SWI/SNF chromatin remodeling complexes.
Regulation of p53 activity through PRMT5-mediated methylation has also been reported, with PRMT5 depletion triggering p53-dependent apoptosis9. In addition, PRMT5 was shown to have regulatory control on the JAK-STAT signaling pathway through methylation of Smad7, enhancing its binding to the IL-6 co-repector gp130 for STAT3 activation and proliferative downstream signalling.10
As mentioned above, histone proteins (specifically H3R8 and H4R3) are also substrates of PRMT5 for symmetrical dimethylation11,12,13, establishing a repressive chromatin environment and limiting the expression of tumor suppressor genes14, such as the RB family of genes15, ST7 and NM2313. Histone H3 K4 acetylation, reflecting an active chromatin environment was also seen to increase upon PRMT5 depletion16.
Of significance in the oncological context, overexpression of PRMT5 and increased SDMA have been established as consequential markers of cancer. For example, PRMT5 was shown to be overexpressed in CRC tissues as compared to surrounding tissue16,17,18, increasing methylation of one of its target proteins, E2F-1 (suppressing its activity and reducing E2F-dependent apoptosis)16. Conversely, low levels of PRMT5 resulting in reduced arginine methylation allows E2F-1 to bind to DNA more efficiently and activate transcription of its target genes, such as those that induce apoptosis16. Importantly, when PRMT5 is depleted in normal cells (experiment used MEFs), it did not induce apoptosis, a therapeutically relevant result in terms of limiting off-target effects16.
In addition to CRC, PRMT5 is also overexpressed in human prostate cancer and correlates with androgen receptor (AR) expression. In this context, PRMT5 acts as an epigenetic activator after its recruitment to the AR promoter and symmetric dimethylation of histone H4R319,20. PRMT5 dysregulation has also been implicated in breast cancer 21, ovarian cancer 22, T and B cell lymphoma/leukemias such as mantle cell lymphoma 23, B-CLL15, lung cancer10, neuroblastoma24, glioma25,26, metastatic melanoma 27, retinoblastoma28, and others, representing a wide range of potential targets for therapeutic intervention.
PRMT5 inhibitors
| Candidate | MoA | Indication | Phase |
|---|---|---|---|
| GSK3326595 (GSK) | Peptide competitive | MDS, AML, Advanced solid tumors (ACC) | I/II |
| JNJ-64619178 (Janssen) | SAM competitive AND peptide competitive | NHL, MDS | I |
| PF-06939999 (Pfizer) | SAM competitive | NSCLC, HNSCC, esophageal, endometrial, cervical, bladder | I |
| PRT543 (Prelude) | SAM competitive | ACC, HRD+ tumors, Myelofibrosis, MDS | I |
| PRT811 (Prelude) | Brain-penetrant SAM competitive | GBM, CNS lymphoma, CNS metastases | I |
| LLY-283 (Eli Lilly) | Brain-penetrant SAM competitive | Gliomas | Preclin |
| JBI-778 (Jubilant) | Brain-penetrant peptide competitive | Glioblastoma, Brain metastases, MCL | Preclin |
| TNG908 (Tango) | MTA cooperative inhibitor | MTAP-deleted cancers | Preclin |
| MRTX9768 (Mirati) | PRMT5-MTA complex inhibitor, MTA competitive | MTAP-deleted cancers | Preclin |
| GSK3235025 (GSK) | Peptide competitive | - | - |
Besides Prelude's candidate, there are three other PRMT5 inhibitors in the clinic: first in class molecule, GSK3326595, JNJ-64619178, and PF 0639999.
GSK3326595
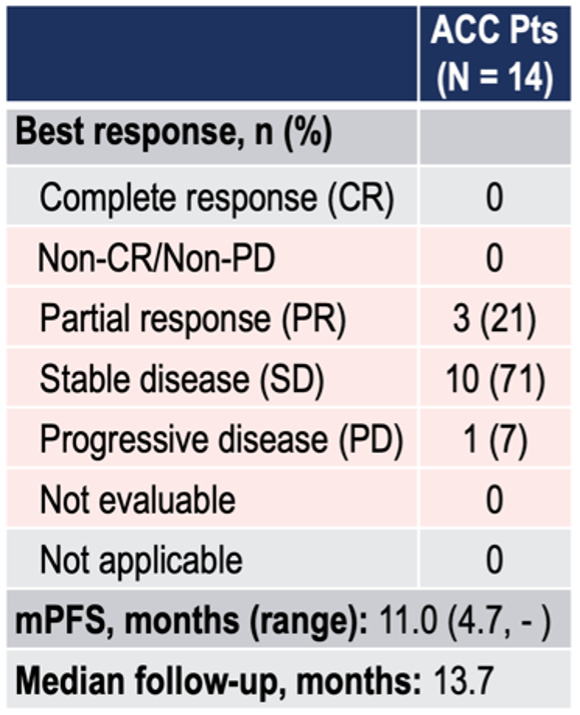
This is the most advanced candidate. It has an IC50 of 6.2 nM and is a substrate/peptide competitive inhibitor. The three-part Phase I trial is ongoing and recruiting (METEOR-1 study, NCT02783300), with a planned total enrollment of 322 patients. Part 1 (dose escalation portion) of the trial has closed with data presented at ESMO 2019, as below. Notably, of 14 ACC patients, 3/14 had PRs, and 10/14 had stable disease.
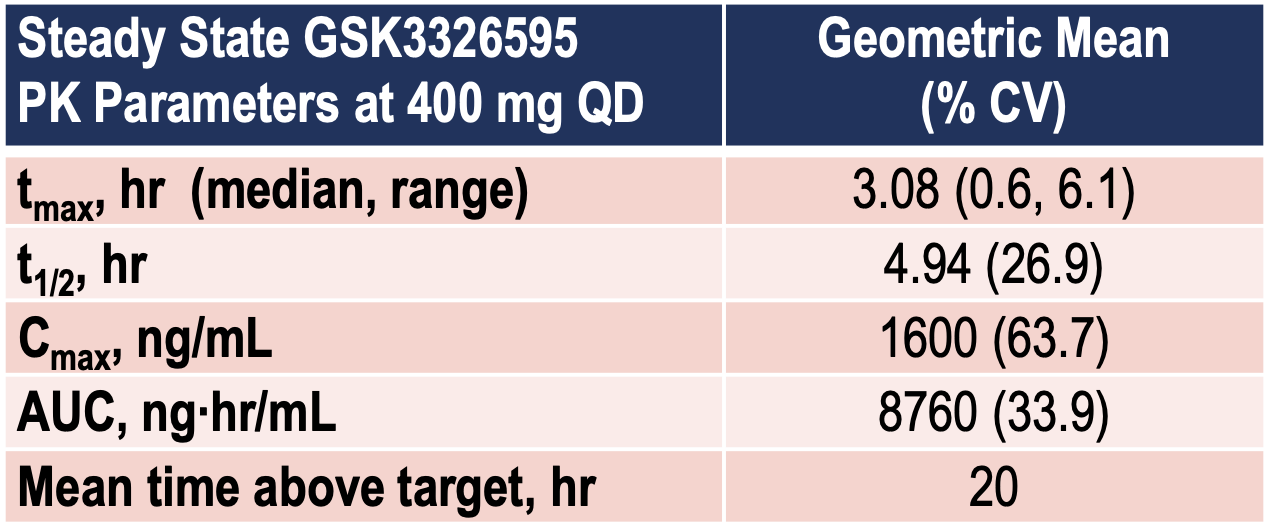
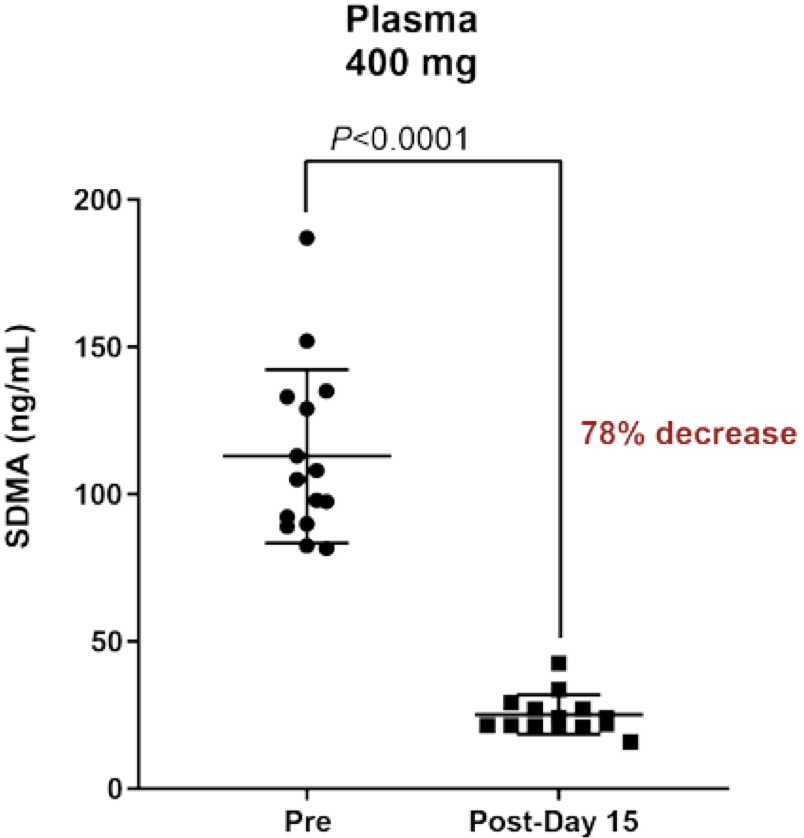
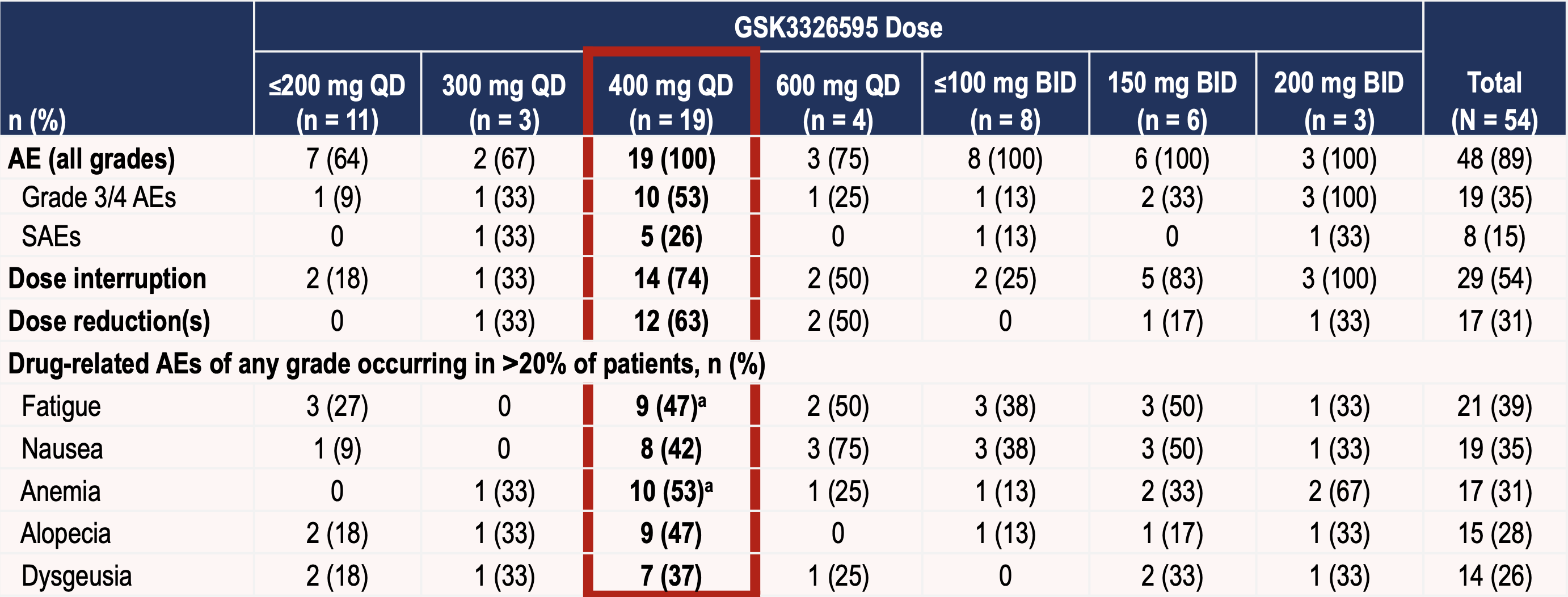
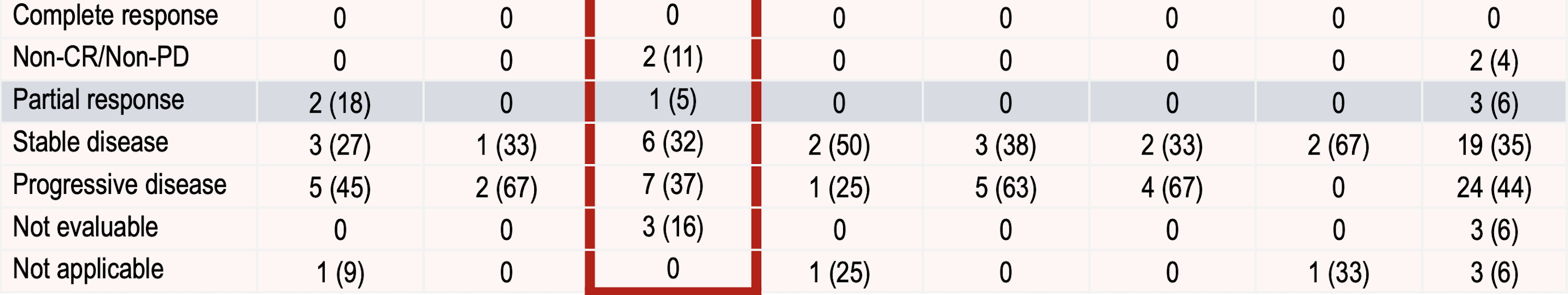
A Phase I/II study in MDS and AML is also ongoing. See ASH 2019 presentation for the study design of that trial here.
JNJ-64619178
A SAM-competitive and peptide-competitive PRMT5 inhibitor (binds simultaneously to the SAM and protein substrate binding pockets of the PRMT5-MEP50 complex). A Phase I trial (NCT03573310) is ongoing in solid tumors, NHL and MDS. The drug demonstrated efficacy in several NSCLC/SCLC xenograft models. Initial data from 54 patients in the phase I trial was presented at ESMO 2020 that can be viewed here.
Of Grade 3/4 TRAEs, 20% of patients had thrombocytopenia, 17% had anemia, 6 % neutropenia. 30/54 patients had dose interruptions or reductions due to AE.
From 11 ACC patients, a confirmed PR was observed. Of patients with ACC, prostate cancer, salivary gland carcinomas and other tumor types, 7 had SD for >6 months.
PF-0639999
A SAM competitive inhibitor. Of the clinical PRMT5 inhibitors, its binding site is likely closest to that of PRT543. The drug is in Phase I (NCT03854227) in patients with NSCLC, HNSCC, esophageal, endometrial, cervical and bladder cancers. The trial is currently enrolling in dose expansion cohorts. Initial results were presented at ASCO 2021 here.
The most common Gr3+ TRAEs were anemia (25%) thrombocytopenia (21%), fatigue, neutropenia and lymphocyte count decrease (4% each). Plasma SDMA was reduced at steady-state (58.4-87.5%), similar to GSK's compound.
From 23 patients, there were 2 PRs, one in HNSCC and one in NSCLC, and 9 SDs. The recommended monotherapy dose selected was 6 mg QD.
LLY-283
A brain-penetrant SAM-competitive PRMT5 inhibitor. In a recently published article, it was shown to suppress growth of 46 patient-derived GBM stem cell cultures26
JBI-778
A brain-penetrant substrate-competitive inhibitor. A recent presentation at AACR 2021 (poster #1128) showed tumor growth inhibition in an orthotopic glioblastoma model. The company is aiming to file an IND in 1H22.
TNG908
An MTA cooperative inhibitor that leverages the accumulation of MTA in MTAP-deleted cancers to inhibit PRMT5. This compound is being developed by Tango Therapeutics to take advantage of the PRMT5-MTAP synthetic lethal pair for a potentially effective therapy with limited off-target effects.
The company recently went public through a SPAC merger. They plan to file an IND for TNG908 in Q4 of this year and initiate a Phase I/II trial in 1H21.
MRTX9768
Mirati is also getting into the space, with its PRMT5-MTA complex selective inhibitor, specifically for methylthioadenosine phosphorylase (MTAP)-deleted cancers (about 10% of all cancers). MTA (methylthioadenosine) is an analogue of SAM and competes with it for the PRMT5 catalytic site as an inhibitory co-factor. In tumors with CDKN2A/MTAP deletion, a build up of MTA occurs resulting in inhibited PRMT5 activity and reduced levels of SDMA29, However, a resulting PRMT5-dependency confers increased sensitivity to PRMT5 depletion30,31, making it a viable target for restoring susceptibility to apoptotic inducing factors. This vulnerability extends to the upstream enzyme MAT2A29 (of which Ideaya Biosciences and others are targeting).
MRTX9768 is the first PRMT5 inhibitor to target the PRMT5-MTA complex. The company is planning to file an IND in 1H22.
GSK3235025/EPZ0156666
A SAM-uncompetitive, peptide competitive inhibitor discovered at Epizyme and can be found in the literature, but has not been taken forward into clinical development. Epizyme's methyltransferase inhibitors were licensed to GSK in a $650 M deal ($20 M upfront + $630 M milestones) in January 2011. I suspect GSK chose to focus on '6595. In 2015, a publication showed in vivo efficacy in MCL models32. A recent publication28 showed '5025 could inhibit retinoblastoma cell proliferation and showed in vivo efficacy in a xenograft model.
Closely-related competition
MAT2A inhibitors can reduce PRMT5 methylation activity and PRMT5-dependent mRNA splicing 33 by reducing synthesis of SAM34. Some of these may be useful to keep an eye on, such as IDE397 (Phase I, IDEAYA Biosciences) and AG-270 (Phase I, Servier).
PRT543 & Data
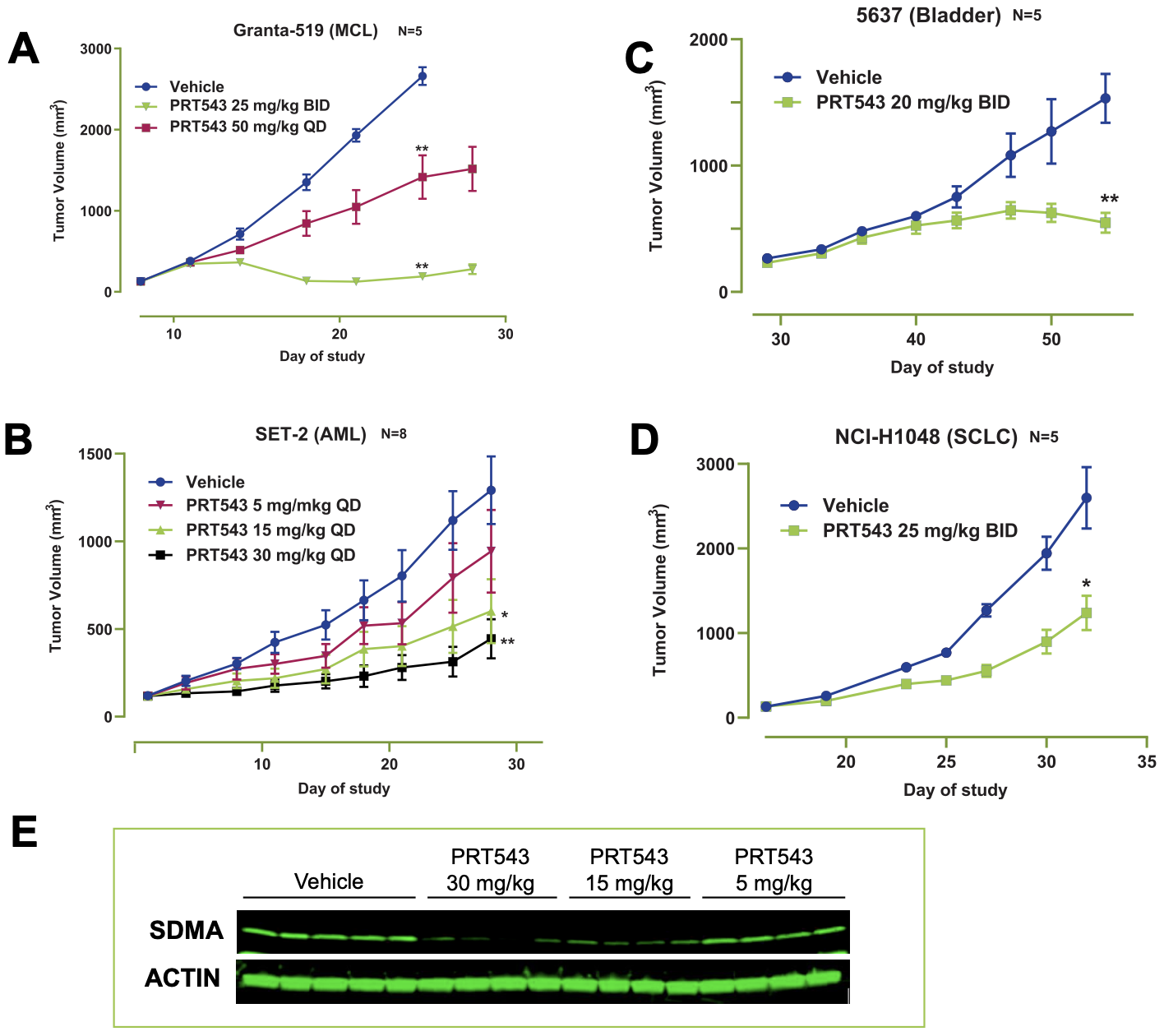
PRT543 is a SAM competitive inhibitor with an IC50 (PRMT5) of 10.8 nM. Pre-clinical data was presented at AACR 2020 (Poster #2915) which can be viewed here.
Oral administration of PRT543 showed tumor growth inhibition in multiple xenograft models, including those derived from MCL (mantle cell lymphoma), bladder, AML and SCLC cell lines. This correlated with reductions in SDMA.
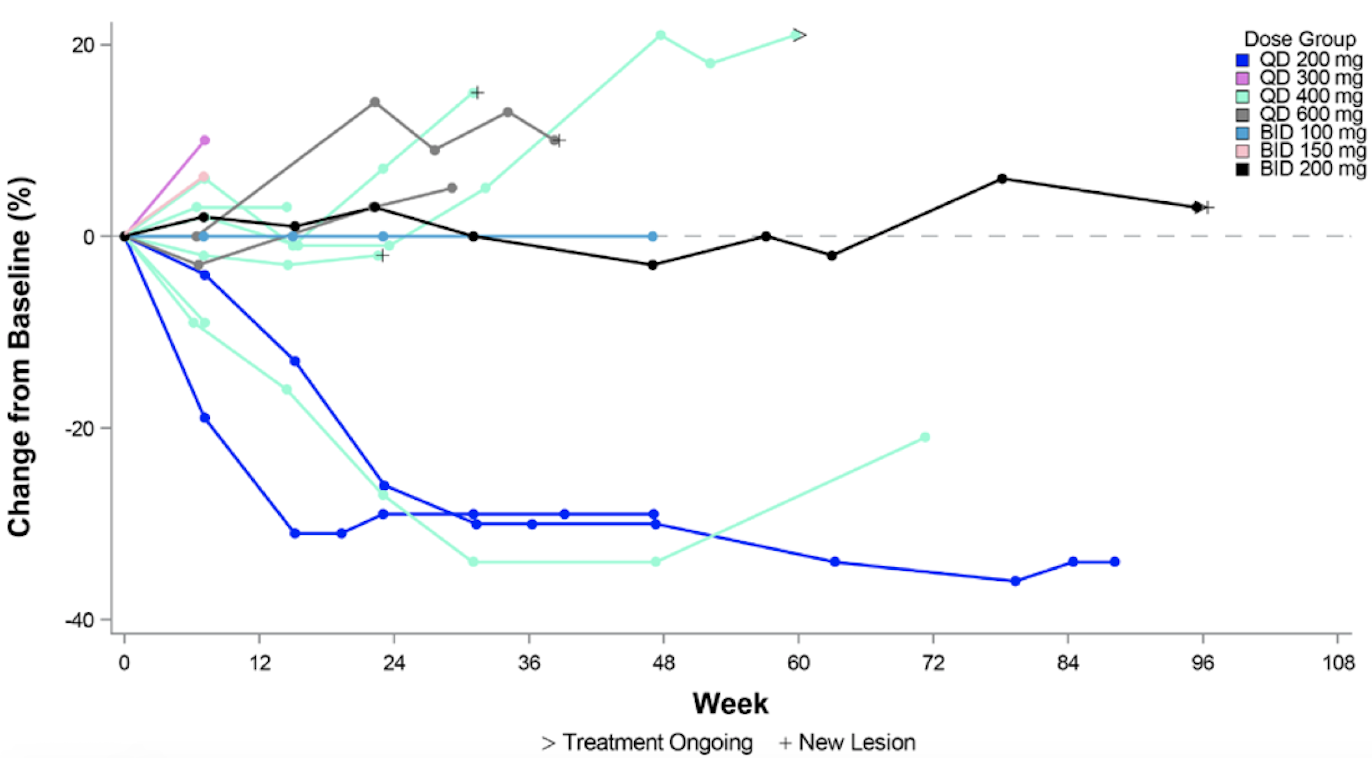
The Phase I clinical trial was initiated in Feb 2019 and has two groups, Group A (Solid tumors + NHL) and Group B (MDS and myelofibrosis). After dose escalation, the expansion phase will include cohorts from Group A of n=40 with ACC, n=40 with HRD+ solid tumors, n=20 with spliceosome-mutated solid tumors and n=40 with Myelofibrosis/MDS, n=20 combination with ruxolitinib in myelofibrosis, n=20 myeloid malignancies with spliceosome mutations from Group B. Prelude is selecting these tumour types on the basis that they may be potentially driven by PRMT5 dysregulation.
See interim Phase I PK/PD data below.
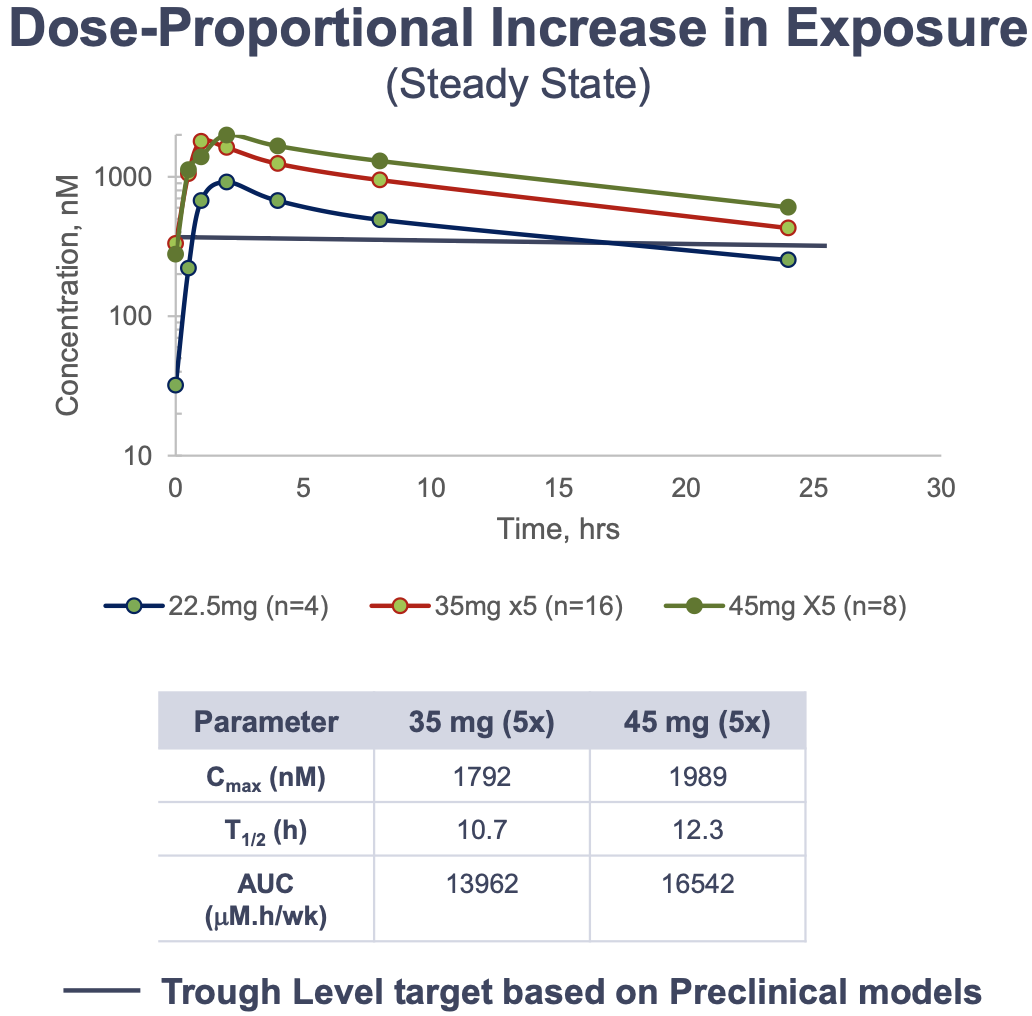

As seen with the other clinical PRMT5 inhibitors, thrombocytopenia and anemia remain to be the most common Grade 3/4 TRAEs. Of 61 patients enrolled in the trial (42 with solid tumors including 2 HRD+ high grade serous ovarian cancer, 11 with myelofibrosis, 7 with MDS and 1 with NHL), there were 24 SAEs reported from 11 patients, 3 of which were deemed drug-related. Importantly, there were no patient discontinuations related to AEs, and thrombocytopenia remains the only DLT.
The mean reduction in SDMA was approximately 75% at both the 35 mg QD and 50 mg QD doses representing maximum inhibition of PRMT5 activity.
On the efficacy side, a CR was confirmed in the HRD+ high grade serous ovarian cancer patient (the first enrolled biomarker positive patient). The patient had 7 prior lines of therapy including PARPi and had one target lesion lymph node at baseline measuring 19mm. Time to response was quick, with a CR as per RECIST v1.1 determined at the first follow up tumor assessment (8 weeks after enrollment, regression in size of the target lesion to 8 mm). The target lesion regressed in size to 5 mm by the second follow-up (week 16) and remained at 5 mm at the third follow-up (week 24), demonstrating durability of the response. The patient has received 9 months of therapy and has remained in CR (as of December 16 2020).
In Group B, 11 patients were enrolled, (9 MF and 2 MDS). One MF patient receiving 20 mg twice bi-weekly had an objective response of clinical improvement and has been on treatment for over a year. Another MF patient receiving 40 mg three times weekly demonstrated a 66% decrease in TSS. Eight patients achieved a best response of SD. Two patients remained on study for approximately one year.
Over 60 patients have been enrolled in the Phase I trial to date and the trial is in its dose expansion portion. The company is presenting data from the dose escalation portion at the AACR-NCI-EORTC triple meeting in October.
Prelude's position
Of the four companies with PRMT5 inhibitors in clinical trials, Prelude is the only SMID-cap. PRT543 is wholly-owned, and may represent an opportunity for a large-cap partner to get in the space, such as Roche or AstraZeneca. Incyte could also be interested for the potential combinatory benefit in myelofibrosis and close ties to management.
There are ~15,000 patients with ACC in the US with ~1,200 new diagnoses a year (according to ACCoi.org). The 5 and 15 year survival are 89 and 40% respectively, (according to cancer.net). Patients are typically initially treated with surgical resection followed by radiation. Advanced/metastatic disease is treated with chemotherapy and TKIs with generally low response rates and durability. This is an area of unmet need.
HRD+ tumors are typically treated with PARP inhibitors and have generated over $1.6 billion of revenue in 2019 (PRLD 2021 10-K). There are currently no options for patients after PARP inhibitors. Ruxolitinib (Incyte) revenues in MF were $1.6 billion in 2019.
There are about 1800-2500 patients diagnosed with uveal melanoma in the US per year35, with no treatments FDA approved for advanced disease (very likely changing soon with Immunocore's tebentafusp (for HLA-A*02:01 patients only).
PRT811
This is a PRMT5 inhibitor that has demonstrated brain-penetrance, which Prelude is testing for glioblastoma and other CNS metastatic cancers. It is the only clinical-stage PRMT5 inhibitor with reported brain penetrance, but will face competition soon from Lilly and Jubilant.
A poster (#2919) presented at AACR 2020 can be viewed here. They showed brain penetrance in mice and rats and tumor inhibition in a GBM xenograft model.
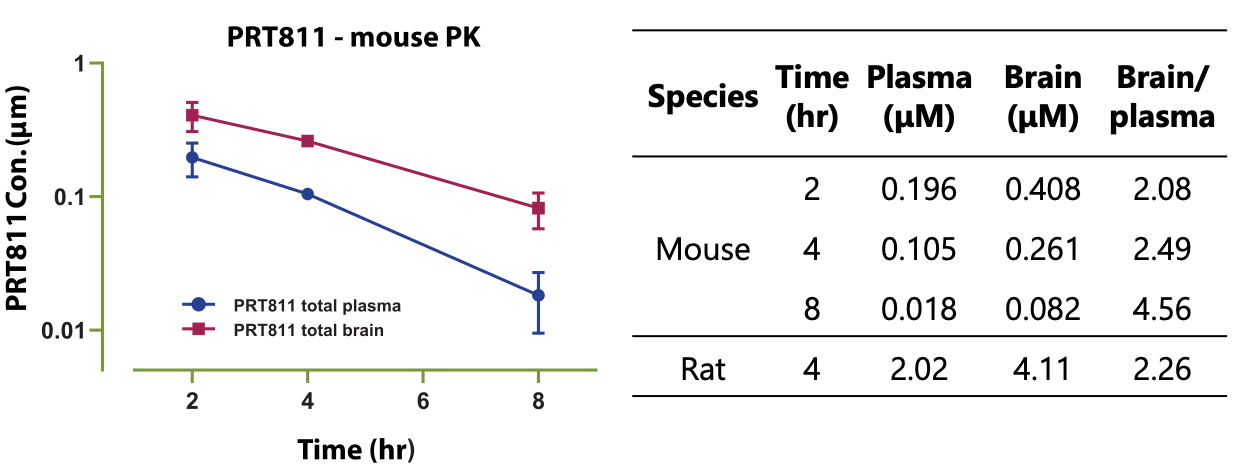
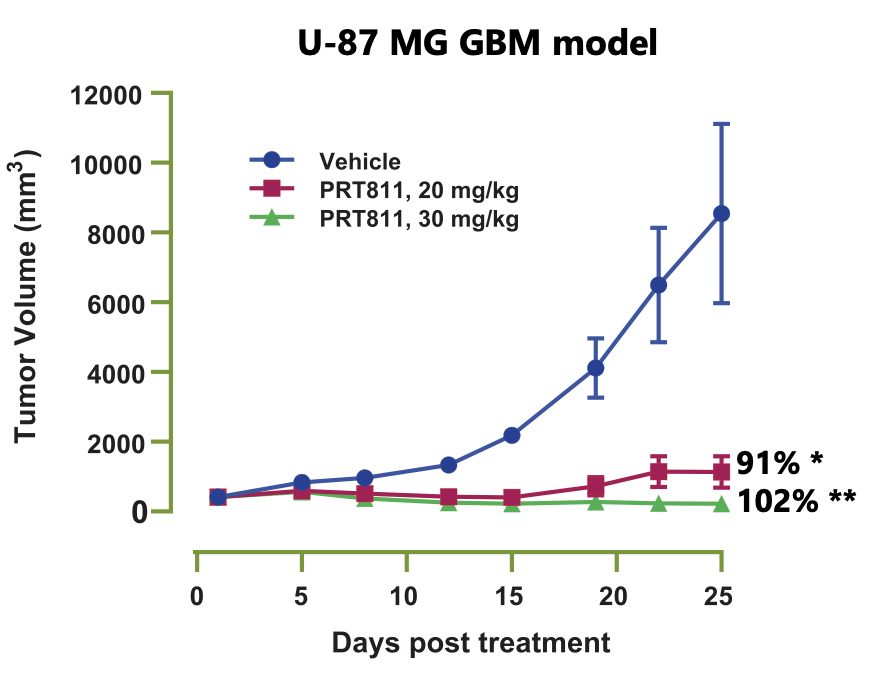
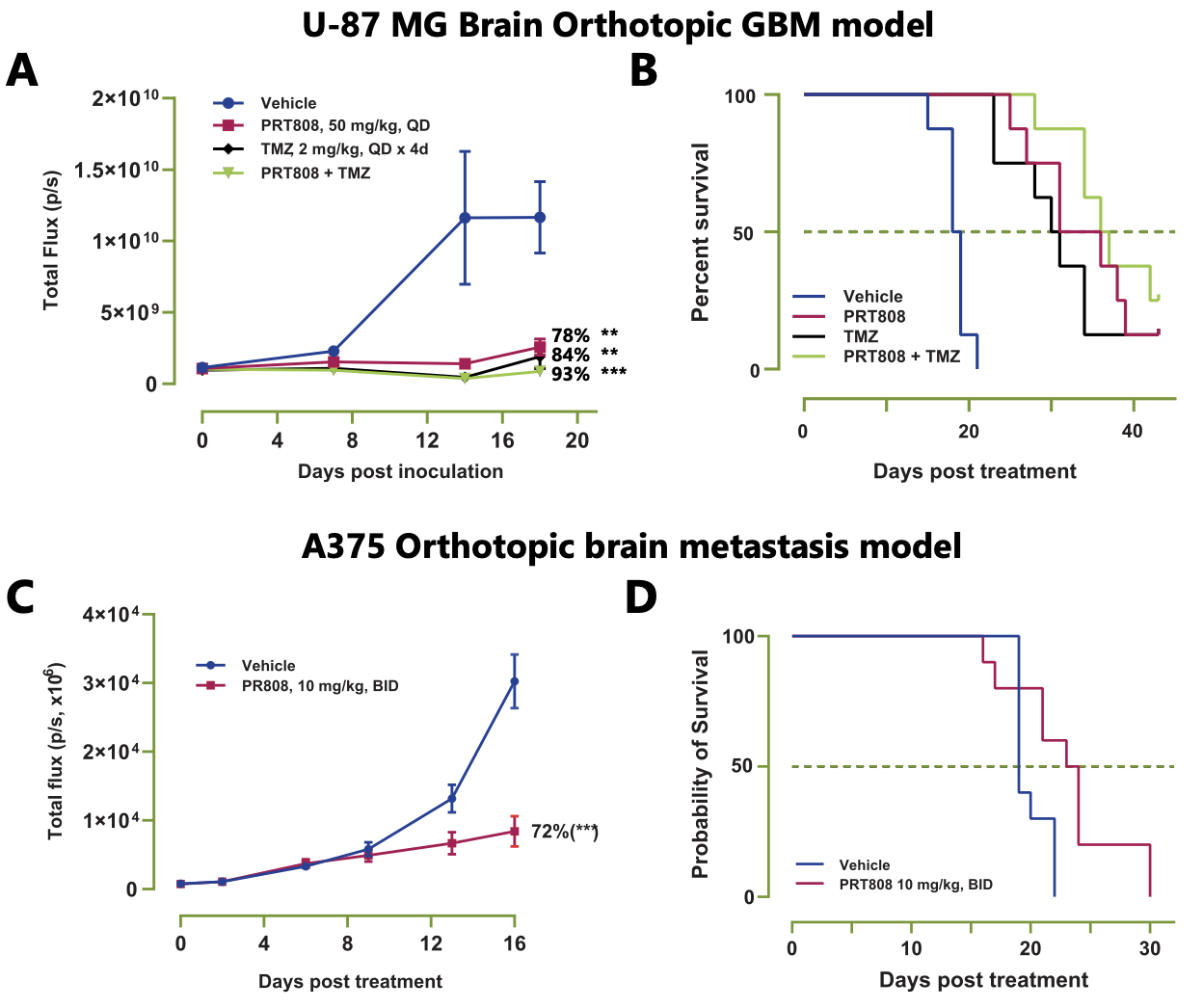
The Phase I is currently enrolling in the planned dose expansion cohorts (n=20 glioblastoma, n=20 Primary CNS lymphoma, n=20 CNS metastatic disease) with initial clinical data being presented at AACR-NCI-EORTC.
Interim data of 24 enrolled patients (16 with advanced solid tumors and 8 with GBM) showed no SAEs deemed related to treatment and no DLTs. However, one patient discontinued due to transient Grade 2 nausea.
There was one confirmed PR in patient with GBM. The patient entered the trial with progressive disease and one target lesion measuring 23 mm x 10 mm (IDH+, MGMT unmethylated) and was dosed 200 mg (QD two weeks on/one week off). At the first follow-up, a 66% regression in size of the lesion was seen. At the second follow-up (week 18), a partial response was confirmed as per RANO criteria with a 77% regression in lesion size. As of December 16, 2020, the patient received 5 months of therapy and remained in PR with clinical stability.
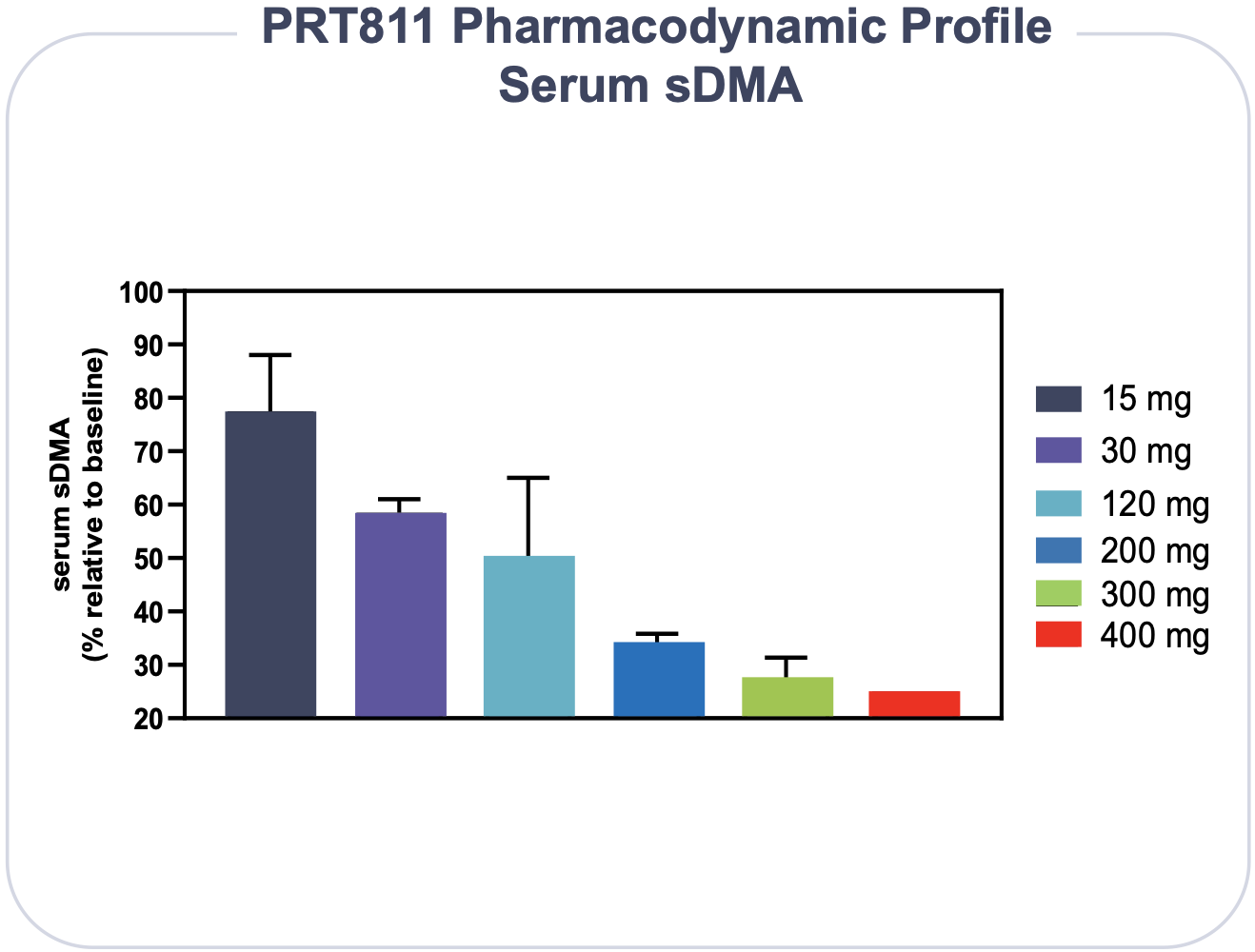
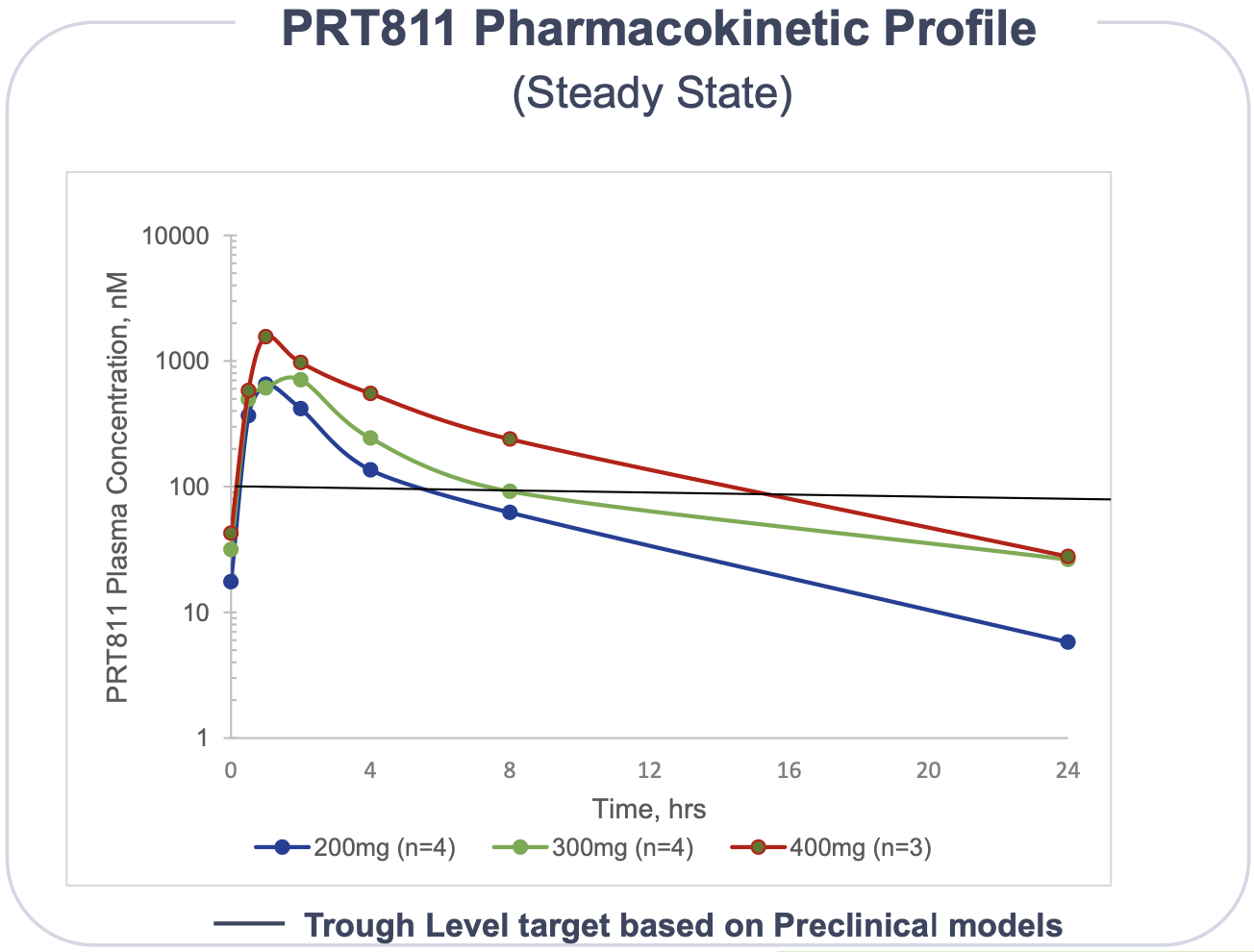
PRT811 Opportunity
There are 14,000 new diagnoses of glioblastoma multiforme in the US per year (according to cancer.gov). Standard of care is typically surgical resection followed by concurrent radiotherapy and temozolomide (TMZ). Tumors with methylation at the MGMT promoter (resulting in epigenetic silencing of the DNA repair enzyme, O6-methylguanine–DNA methyltransferase, occuring in about 35-45% of patients36,37, are significantly more responsive to TMZ with better prognoses36,37. Conversely, treatment with TMZ does not improve survival in patients with MGMT-unmethylated glioblastomas (55-65% of patients)38 making up a large patient group with unmet needs. In one trial, a median OS of 21.7 vs 12.7 months was reported36.
TMZ is a alkylating agent that methylates guanine at N7 and O6, causing nicks in DNA and leading to cell cycle arrest and apoptosis, of which the MGMT enzyme can repair if present and active. While poor responses in patients with unmethylated-MGMT is related to TMZ's mechanism of action, it reveals two things about these tumors: high probability of dysregulated methylation and a need for new therapies. It is also possible that a TMZ/PRMT5i combination in this context could induce synthetic lethality.
A recent paper39 also demonstrated dysregulation of splicing machinery as another major prognostic factor for survival in glioblastoma patients. They highlighted dysregulated SRSF3 (Serine and Arginine Rich Splicing Factor 3, a member of the SR family involved in spliceosome activity) in particular, but also other RNPs, such as PTBP1 and RBM3. Silencing of SRSF3 with siRNAs in a xenograft model was shown to significantly reduce tumor cell proliferation, tumor volume and induce apoptosis. Note that this dysregulation is also prominent in other cancer types, such as prostate cancer40. These data further support the testing of PRMT5 inhibitors in glioblastomas.
Prelude is also targeting primary CNS lymphoma and a subset of metastatic CNS patients with sensitivity to PRMT5 inhibition. There are 1,500 new cases of primary CNS lymphoma in the US per year (according to rarediseases.org), with about 30% refractory to primary treatment41, and 200,000 CNS metastatic patients annually (Prelude presentation).
PRT1419
The Intrinsic Pathway of Apoptosis
The Bcl-2 family of proteins
The Bcl-2 family of proteins share conserved protein domains called Bcl-2 homology (BH) domains. They can be subdivided into three types:
1. BH3-only proteins, pro-apoptotic members containing only BH domain 3. Their BH3 domain is exposed permanently. These proteins bind transiently to pro-apoptotic effector proteins to 'activate' them, triggering the conformational changes needed for mitochondrial outer membrane pore formation. Their expression is tightly regulated.
2. Pro-survival proteins (such as BCL-2, BCL-XL and MCL-1). They have all four BH domains. Their significance is explained further below.
3. Pro-apoptotic effector proteins (Bak and Bax), which homo-oligomerise, cluster and form pores on the mitochondrial outer membrane, resulting in cytochrome c release, caspase activation and cell death42. They have all four BH domains, and their BH3 domain is buried in the inactivated conformation.
Pro-survival proteins
The "pro-survival" proteins are structurally similar to Bak and Bax, sharing all four BH domains, but they do not undergo the conformational changes that lead to oligomerisation (formation of BH3:groove dimers, which cluster and form pores on the mitochondrial outer membrane)43,44,45. Instead, they act as a BH3 "sink", capturing any exposed BH3 domains of BH3-only proteins and Bax, as well as the transiently exposed BH3 domains of activated Bak and Bax46. These two binding functions inhibit apoptosis and are referred to as sequestration by MODE-1 (direct inhibition of BH3-only proteins) and MODE-2 (inhibition by heterodimerization with Bak/Bax)46.
The intrinsic (or, mitochondrial) pathway of apoptosis is regulated in an intricate balance between the pro-survival proteins and BH3-only proteins. In physiological conditions, DNA damage leading to activation of ATM/ATR kinases47 and transcription factors such as p53 lead to the upregulation of BH3-only proteins, tipping the balance of the Bcl-2 family of proteins to favor apoptosis. However, in dysregulated conditions such as those in genomically unstable cancers, pro-survival proteins, such as Bcl-2, Bcl-xL and/or Mcl-1 are overexpressed, leading to MODE-1 and MODE-2 sequestration and evasion of apoptosis. This tumor resistance mechanism poses a signifcant challenge in clinical treatment.
The BH3-mimetic (or, Bcl-2 inhibitor), venetoclax, acts to shift the balance of proteins in favour of apoptosis. However, upregulation of other (untargeted) members of the Bcl-2 family of proteins poses a problem for this mechanism of action, and a major cause of refractory/relapsing cancer48 For example, 21% of r/r CLL patients in clinical trials did not respond to venetoclax49, with strong indications that resistance is due to the presence of pro-survival proteins other than Bcl-250
Venclexta, and beyond Bcl-2 inhibition
Finding new targets in the intrinsic apoptotic pathway has been a hot space since the huge success of venetoclax (Venclexta).
Venclexta is currently FDA-approved for the treatment of CLL/SLL, and newly diagnosed AML (ages 75+ or patients ineligible for intensive induction chemotherapy). It's initial approval (for patients with 17p deletion CLL who have had at least 1 prior therapy) was in 2016, with full approval gained for CLL/SLL in June 2018. Its use today in this setting is mainly in the second and third line of therapy, after chemotherapy and ibrutinib. It received approval for AML in November 2018, and its use in this setting is growing. Venclexta generated a total of $1.3 B in sales in 2020, and is in numerous clinical trials (combinations+monotherapy) for the treatment of other hematological malignancies.
Targeting MCL-1
MCL-1 is another key pro-survival protein, and its overexpression has been well documented as a tumor resistance mechanism to therapies like venetoclax.
For example, MCL-1 is the most dominant pro-survival protein in multiple myeloma (MM). About ~40% of MM patients carry a gain or amplification of 1q21, resulting in upregulation of MCL-151. Venetoclax was only shown to be effective in a subset of MM patients, particularly those with a translocation at t(11;14), where an increased BCL2/MCL1 ratio results in BCL-2 dependence52. MCL-1 expression is also consistently high in AML, unlike Bcl-2 and Bcl-xl, where expression is seen to be more variable53.
Additionally, MCL-1 inhibitors have been shown to work synergistically with BCL-2 inhibitors in preclinical models, rescuing efficacy in venetoclax-resistant relapsing AML54,55,56.
For more, and a recent review discussing targeting MCL-1 in hematological malignancies, I recommend this paper.
Safety & The Cardiac Issue
MCL-1 has an essential role in cardiomyocytic homeostasis, particularly in the induction of autophagy57,58. The potential of cardiotoxicity from MCL-1 inhibitors has been recognised for some time.
For example, deletion of MCL-1 in a mouse model was shown to result in severe contractile dysfunction and death57,58. However, these effects are the result of complete knockdown of MCL-1, and do not represent the degree of inhibition that would occur from drug administration. Perhaps most importantly - these are rodents, and not perfect models. Importantly, Mcl-1+/- heterozygous mice with a 50% reduction in MCL-1 expression showed no impact on general health and no significant differences (in terms of toxicity) to wild type mice when treated with a range of chemotherapeutics59.
Nonetheless, given these data, special attention should be given to determining the therapeutic window in clinical trials. It may be critical to maintain drug exposure below certain thresholds, which will naturally favor some drugs over others depending on their pharmacokinetic profile.
MCL-1 Inhibitors: competition & real-world experience
| Company | Candidate | Indication, Administration | Phase |
|---|---|---|---|
| Novartis & Servier | MIK665 /S64315 | DLBCL, MM, AML/MDS ± BCL-2 inhibitor, ± Azacitidine - IV | I/II |
| AstraZeneca | AZD5991 | AML ± Venetoclax - IV | I/II |
| Amgen | AMG 176 | MM, AML - IV | I |
| Prelude | PRT1419 | MM, AML, NHL, MDS - ORAL, Solid tumors - IV | I |
| Ascentage Pharma | AS00491 | - IV | Preclin |
| Ascentage Pharma | APG-3526 | - IV | Preclin |
| Discontinued | |||
| Amgen | AMG 397 | AML, MDS, lymphoma - ORAL | I |
| AbbVie | ABBV467 | MM - IV | I |
AMG 176
Currently recruiting patients in a Phase I trial for r/r MM and r/r AML (NCT02675452). The trial had been put on voluntary clinical hold for some time after a cardiotoxicity signal was reported with Amgen's other MCL-1 inhibitor, AMG 397.
Another phase I trial (NCT03797261), a combination study with venetoclax for patients with r/r AML, NHL/DLBCL was not restarted.
Preliminary data in patients with r/r MM was released in 201960. At the data cut-off date (March 15, 2019), 26 patients had received the drug with a median of 2 cycles (1-8). Patients had a median of 5 prior lines of therapy, with 20 patients (77%) having received >=5 prior lines of therapy. Grade 3+ TEAEs occured in 16 patients, 9 with neutropenia (35%), 4 anemia (15%), 2 hypertension (8%), and 1 other unspecified. There were two fatal AEs, one of which was treatment related (tumor lysis syndrome), and one hepatic failure (related to disease progression). 11 patients had SD as best overall response. Most patients (22, or 85%) discontinued due to progressive disease.
AZD5991
A Phase I/Ib/IIa 3-part study (NCT03218683) is currently recruiting patients with r/r AML to be treated with AZD5991 as monotherapy or in combination with venetoclax. The study began in August 2017. A lot of pre-clinical data has been presented, but none yet from the clinical trial.
MIK665/S64315
Being developed in a Servier-Novartis partnership. Two Phase I trials, one in r/r AML/MDS (n=38) and another in r/r MM & DLBCL (n=31), have been completed (in May 2020 and June 2019 respectively). The drug is currently in three other Phase I trials for patients with AML, testing combinations with venetoclax (NCT03672695, initiated November 2018), azacitidine (NCT04629443, initiated February 2021) and BCL-2 inhibitor VOB560 (NCT04702425, initiated June 2021, also recruiting NHL and MM patients).
AMG 397
This is the only other orally-administered MCL-1 inhibitor to reach the clinic besides Prelude's. In September 2019, the Phase I trial (NCT03465540) was placed on clinical hold after a cardiac toxicity signal. No further details were given and data has not been released from this trial. The drug has since been discontinued, with Amgen shifting their focus to development of AMG 176.
ABBV467
A first in human Phase I trial was initiated in May 2020, but AbbVie terminated the trial shortly thereafter, citing strategic considerations. Only 8 patients were enrolled.
PRT1419 & Data
Prelude's drug is highly soluble with high oral bioavailability. It is a BIM-competitive inhibitor. It is the only orally-administered MCL-1 inhibitor in clinical trials.
Preclinical work showed robust activity with once-weekly dosing. Importantly, there has been no preclinical evidence of cardiac toxicity.
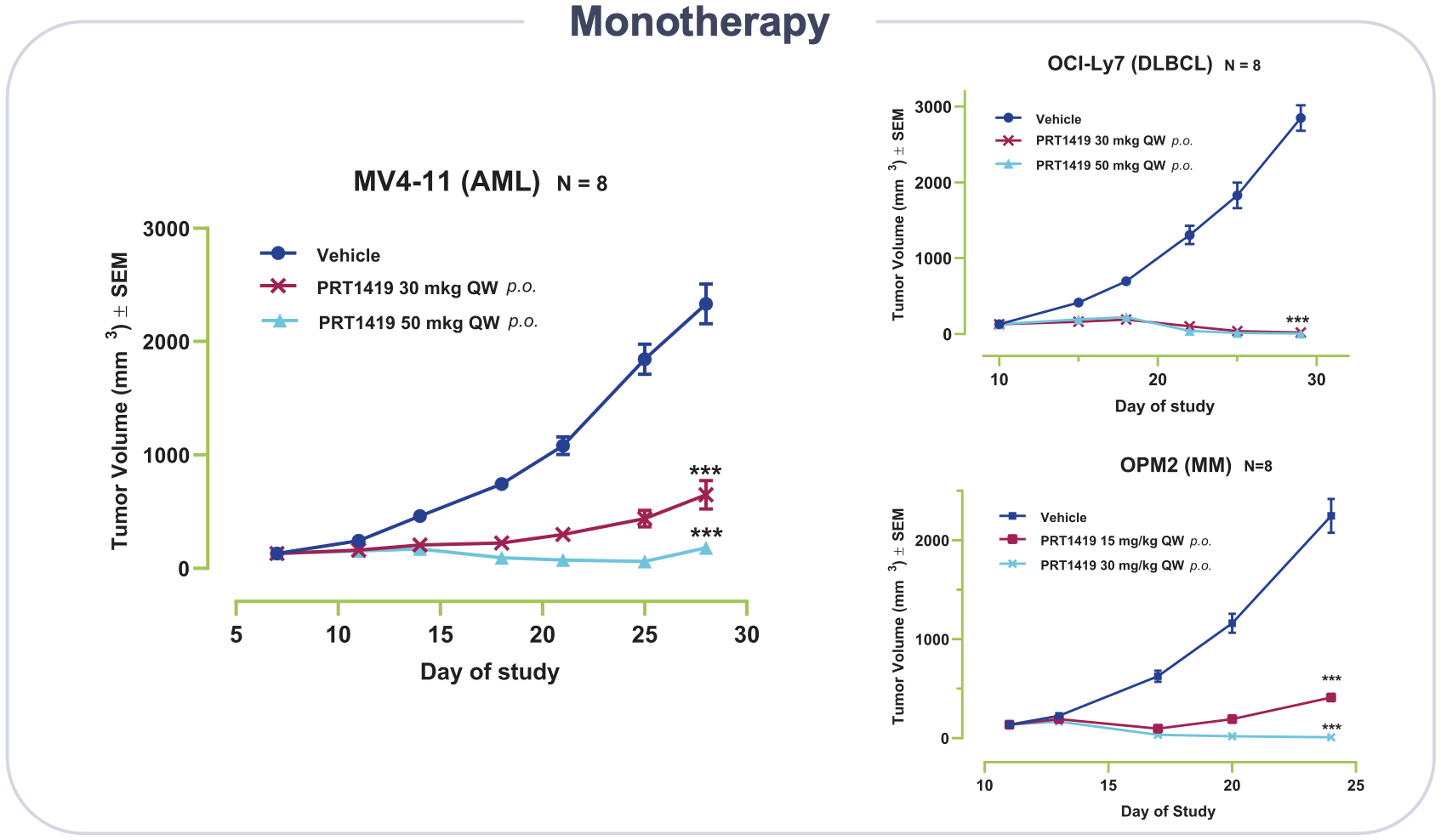
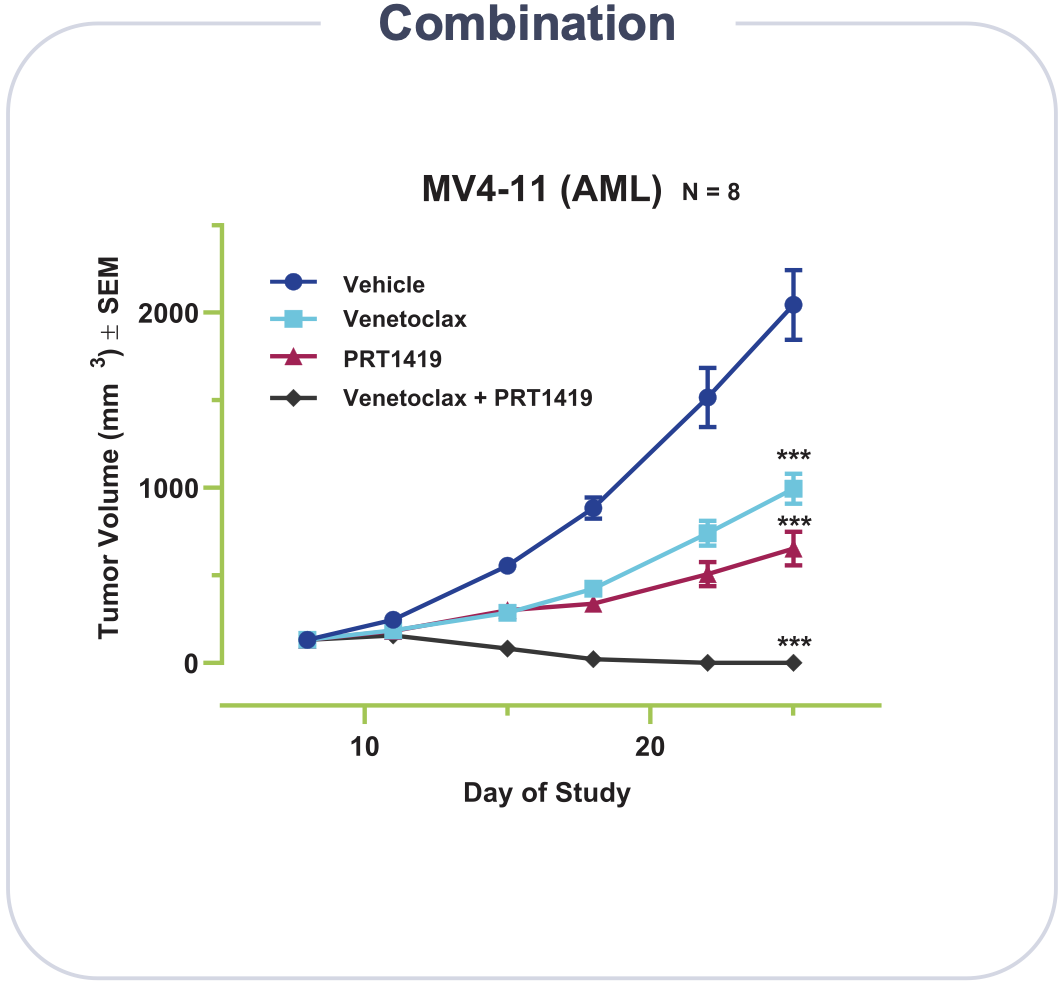
The Phase I study will dose escalate in two groups, Group A (AML and/or high-risk MDS) and Group B (NHL/MM). The expansion phase will test PRT1419 as monotherapy or in combination with either azacitidine or venetoclax in a subgroup of AML/MDS patients.
The first patient was dosed in September 2020. A small clinical update was released some months after, reporting no AEs above Grade 1/2 from 4 enrolled patients (as of December 16, 2020). Dose escalation is currently ongoing.
Prelude is also planning to test an intravenous formulation of the drug in advanced solid tumors. The IND was cleared in March 2021. The trial (NCT04837677) is anticipated to recruit patients with sarcoma, melanoma, lung and breast cancer, and is set to initiate sometime in the near future.
Closely-related competition
CDK9 inhibitors - these drugs act upstream by inhibiting transcription of MCL-1. There is strong evidence to suggest CDK9 and MCL-1 are closely linked. For more, please refer to the VINC report for an overview of CDK9 inhibition and inhibitors in clinical development. Note that Prelude also has a CDK9 inhibitor in development.
PRT2527
CDK9 inhibition
CDK9 forms a complex with a cyclin binding partner (typically Cyclin T1) to positively regulate transcription elongation (through phosphorylation of RNA Pol II). Its activity is significantly upregulated in various cancer types resulting in high expression of proliferation/survival proteins such as MYC and MCL-1. CDK9 inhibition has been shown to reduce MYC mRNA and MCL1 mRNA.
CDK9 competition
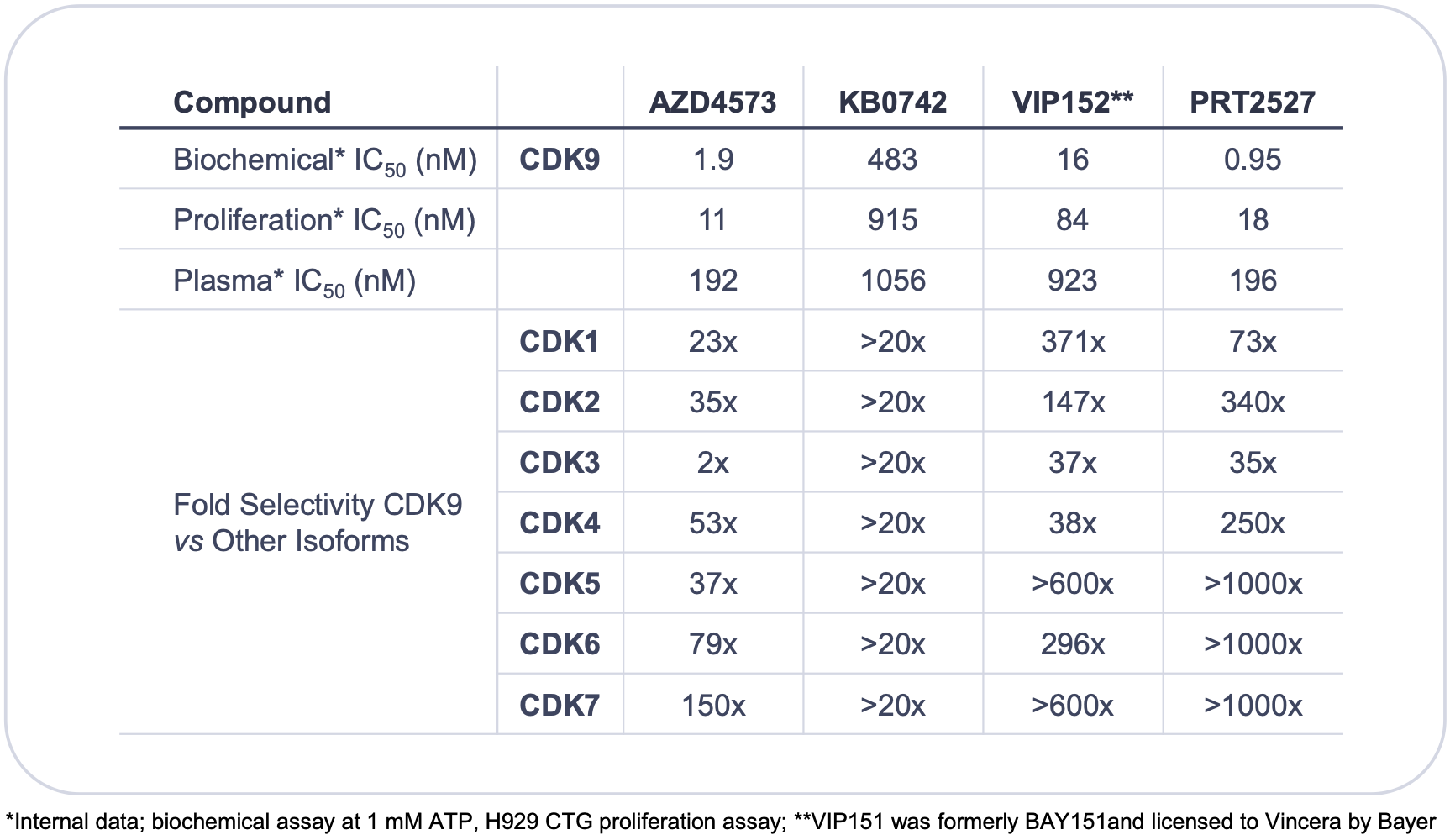
Refer to the VINC report for more details. The most advanced candidate is AstraZeneca's AZD4573. The issue with old CDK9 inhibitors was a lack of selectivity resulting in a low therapeutic index and poor tolerability. These issues can potentially be circumvented with improved selectivity.
PRT2527 & Data
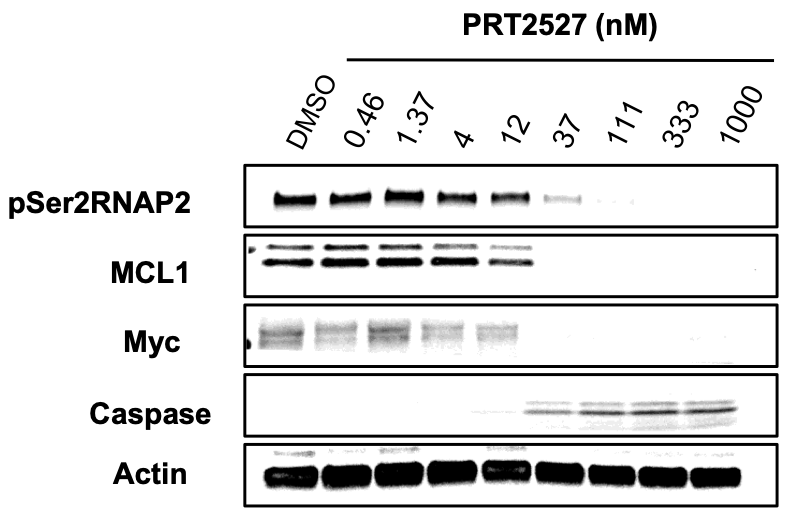
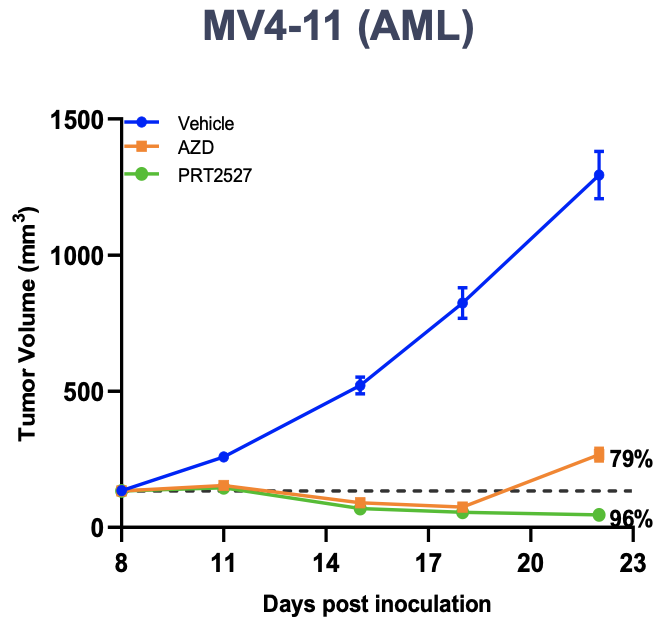
Prelude's CDK9 inhibitor has high selectivity over other CDKs, and may potentially be best-in-class. It is currently in IND-enabling studies with an anticipated IND filing this year. In pre-clinical models, PRT2527 was shown to have high efficacy in a MYC-amplified AML xenograft model at well-tolerated doses.
PRT-SCA2
PRT-SCA2 is Prelude's SMARCA2 degrader currently in IND-enabling studies, with anticipated filing in 2022.
Despite high homology between SMARCA2 and SMARCA4 (~80%), Prelude has discovered a highly-selective SMARCA2 protein degrader (IC50: 0.76 nM), with a >20-fold selectivity over SMARCA4. It targets SMARCA2 for degradation through the E3 ubiquitin ligase pathway. Prelude hopes to use this mechanism to induce apoptosis in SMARCA4-deficient cancer cells in a synthetic lethality approach.
SMARCA2/SMARC4 & Synthetic Lethality
SMARCA2 (also known as BRM) and its closely related paralog, SMARCA4 (also known as BRG1) are ATPase subunits of SWI/SNF (BAF) chromatin remodeling complexes. The SWI/SNF (BAF) complex modulates DNA accessibility to regulate gene expression. It is comprised of either SMARCA2 or SMARCA4, as well as core and accessory subunits.
SMARCA2 and SMARCA4 are frequently mutated in cancer7,8. For example, SMARCA4 mutations are found in ~11% of NSCLC61, of which the majority are homozygous (70-90%) and resulting in the loss of SMARCA4 protein. In this setting, SMARCA2 plays a compensatory role in cells to maintain BAF activity. An important consequence to this is the ability to target these cells through synthetic lethality. Evidence for the synthetic lethal relationship between SMARCA2 and SMARCA4 has been discussed extensively here and here. I recommend reading these papers for more information regarding this space.
An additional but important point to add here is that SMARCA2 is rarely mutated in cancer (except in ACC62, which PRLD is hoping to treat with their PRMT5 inhibitor). The relevance of this is that a wider range of SMARCA4-deficient tumors can be treated, with tumor escape mutants less likely to rise, potentially improving responses and prolonging survival. A synthetic lethal approach to treating SMARCA4-mutant cancers could also provide safety/tolerability benefits by leaving normal cells unharmed.
SMARCA2 degraders
Foghorn Therapeutics has a SMARCA2 degrader in preclinical development.
Ryvu Therapeutics also had a SMARCA2 degrader in research and presented data at AACR 2020, but they have since removed it from their pipeline.
I am not aware of any other SMARCA2 degraders in development.
Patents
As summarised below. All patents are wholly-owned.
| Asset | Patent type & number | Earliest expiry | Jurisdiction |
|---|---|---|---|
| PRT543 | COM (U.S. 10,570,140) | Aug 2038 | USA (ex-US pending) |
| PRT811 | COM (U.S. 10,711,007) | Apr 2039 | USA (ex-US pending) |
| PRT1419 | COM (pending) | Nov 2039 (pending) | - |
| PRT2527 | COM (pending) | 2040 (pending) | - |
| PRT-SCA2 | COM (pending) | - | - |
Finances & Upcoming Milestones
The company believes its current cash and cash equivalents will be sufficient to fund operations into mid 2023 (Q221 press release).
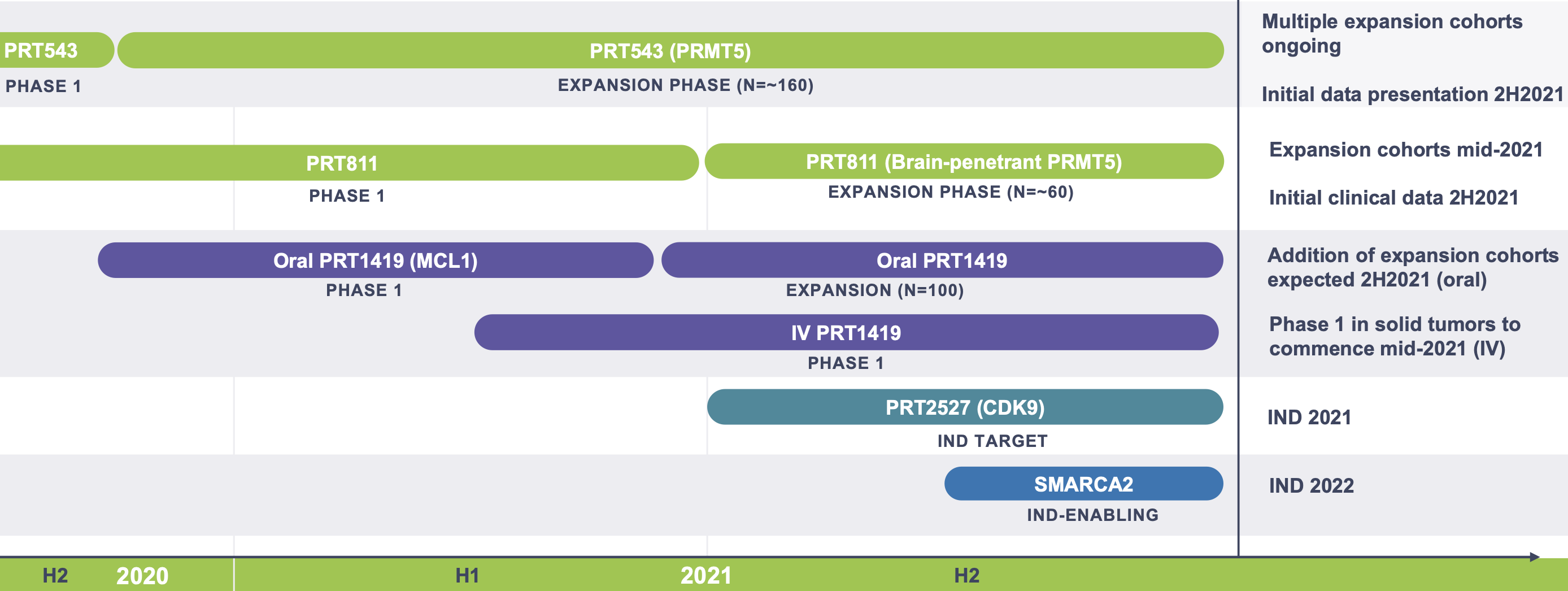
Initial clinical data from the dose escalation portion of the phase I trial of PRT543 and PRT811 will be presented at the AACR-NCI-EORTC triple meeting in October.
Dose escalation of the IV formulation of PRT1419 in solid tumors is underway as of August.
The company is planning to submit an IND application for PRT2527 in 2021 and to begin IND-enabling studies for PRT-SCA2 by the end of the year.
Note: the source of these timelines is Prelude's last quarter press release and subject to change.
Disclosure: Data presented in this article have been obtained from third-party publications and sources. I do not guarantee the accuracy of this data and all information presented here should be checked and verified accordingly. Readers of this article should each make their own evaluation and judgement of the mentioned companies and of the relevance and adequacy of the information provided. Readers should make other such investigation as deemed necessary. The article is intended for informational purposes. Investors and potential investors are requested to do additional research before investing in any of the companies mentioned in this article. All investments have risks of loss associated with them. Investing is very risky, highly speculative, and should not be done by anyone who cannot afford to lose the entire value of investment and without prior due diligence. Investors should evaluate the risks associated with each individual company before investing. All forward-looking statements are anticipations subject to risks and uncertainties and actual results can differ materially from those projected.
Additional disclosure: This article contains trademarks, trade names, copyrights and data of Prelude Therapeutics and other companies, which are the property of their respective owners.
Tweet
References
1. Wang, Y., Hu, W., & Yuan, Y. (2018). Protein Arginine Methyltransferase 5 (PRMT5) as an Anticancer Target and Its Inhibitor Discovery. Journal of Medicinal Chemistry. doi:10.1021/acs.jmedchem.8b0059
2. Antonysamy, S., Bonday, Z., Campbell, R. M., Doyle, B., Druzina, Z., Gheyi, T., … Emtage, S. (2012). Crystal structure of the human PRMT5:MEP50 complex. Proceedings of the National Academy of Sciences, 109(44), 17960–17965. doi:10.1073/pnas.1209814109
3. Hadjikyriacou, A., Yang, Y., Espejo, A., Bedford, M. T., & Clarke, S. G. (2015). Unique Features of Human Protein Arginine Methyltransferase 9 (PRMT9) and Its Substrate RNA Splicing Factor SF3B2. Journal of Biological Chemistry, 290(27), 16723–16743. doi:10.1074/jbc.m115.659433
4. Meister, G., Eggert, C., Bühler, D., Brahms, H., Kambach, C., & Fischer, U. (2001). Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Current Biology, 11(24), 1990–1994. doi:10.1016/s0960-9822(01)00592-9
5. Meister G, Fischer U (2002) Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J 21: 5853–5863
6. Andreu-Perez, P., Esteve-Puig, R., de Torre-Minguela, C., Lopez-Fauqued, M., Bech-Serra, J. J., Tenbaum, S., … Recio, J. A. (2011). Protein Arginine Methyltransferase 5 Regulates ERK1/2 Signal Transduction Amplitude and Cell Fate Through CRAF. Science Signaling, 4(190), ra58–ra58. doi:10.1126/scisignal.2001936
7. Wang, P., Song, X., Cao, D. et al. Oncogene-dependent function of BRG1 in hepatocarcinogenesis. Cell Death Dis 11, 91 (2020). https://doi.org/10.1038/s41419-020-2289-3
8. Pal, S., Yun, R., Datta, A., Lacomis, L., Erdjument-Bromage, H., Kumar, J., … Sif, S. (2003). mSin3A/Histone Deacetylase 2- and PRMT5-Containing Brg1 Complex Is Involved in Transcriptional Repression of the Myc Target Gene cad. Molecular and Cellular Biology, 23(21), 7475–7487. doi:10.1128/mcb.23.21.7475-7487.2003
9. Jansson, M., Durant, S. T., Cho, E.-C., Sheahan, S., Edelmann, M., Kessler, B., & La Thangue, N. B. (2008). Arginine methylation regulates the p53 response. Nature Cell Biology, 10(12), 1431–1439. doi:10.1038/ncb1802
10. Cai, C., Gu, S., Yu, Y., Zhu, Y., Zhang, H., Yuan, B., Shen, L., Yang, B., & Feng, X. H. (2021). PRMT5 Enables Robust STAT3 Activation via Arginine Symmetric Dimethylation of SMAD7. Advanced science (Weinheim, Baden-Wurttemberg, Germany), 8(10), 2003047. https://doi.org/10.1002/advs.202003047
11. Girardot, M., Hirasawa, R., Kacem, S., Fritsch, L., Pontis, J., Kota, S. K., … Feil, R. (2013). PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Research, 42(1), 235–248. doi:10.1093/nar/gkt884
12. Migliori, V., Müller, J., Phalke, S., Low, D., Bezzi, M., Mok, W. C., … Guccione, E. (2012). Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nature Structural & Molecular Biology, 19(2), 136–144. doi:10.1038/nsmb.2209
13. Pal, S., Vishwanath, S. N., Erdjument-Bromage, H., Tempst, P., & Sif, S. (2004). Human SWI/SNF-Associated PRMT5 Methylates Histone H3 Arginine 8 and Negatively Regulates Expression of ST7 and NM23 Tumor Suppressor Genes. Molecular and Cellular Biology, 24(21), 9630–9645. doi:10.1128/mcb.24.21.9630-9645.2004
14. Zhao, Q., Rank, G., Tan, Y. T., Li, H., Moritz, R. L., Simpson, R. J., … Jane, S. M. (2009). PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nature Structural & Molecular Biology, 16(3), 304–311. doi:10.1038/nsmb.1568
15. Wang, L., Pal, S., & Sif, S. (2008). Protein Arginine Methyltransferase 5 Suppresses the Transcription of the RB Family of Tumor Suppressors in Leukemia and Lymphoma Cells. Molecular and Cellular Biology, 28(20), 6262–6277. doi:10.1128/mcb.00923-08
16. Cho, E.-C., Zheng, S., Munro, S., Liu, G., Carr, S. M., Moehlenbrink, J., … Thangue, N. B. L. (2012). Arginine methylation controls growth regulation by E2F-1. The EMBO Journal, 31(7), 1785–1797. doi:10.1038/emboj.2012.17
17. Lim, Y., EunPark, Y., Ha, S., Lee, J. E., & Kim, H. C. (2020). A comprehensive Analysis of Symmetric Arginine Dimethylation in Colorectal Cancer Tissues Using Immunoaffinity Enrichment and Mass Spectrometry. PROTEOMICS, 1900367. doi:10.1002/pmic.201900367
18. Zhang B, Dong S, Zhu R, Hu C, Hou J, Li Y, Zhao Q, Shao X, Bu Q, Li H, Wu Y, Cen X, Zhao Y. Targeting protein arginine methyltransferase 5 inhibits colorectal cancer growth by decreasing arginine methylation of eIF4E and FGFR3. Oncotarget. 2015; 6:22799–811. https://doi. org/10.18632/oncotarget.4332.
19. Deng, X., Shao, G., Zhang, H.-T., Li, C., Zhang, D., Cheng, L., … Hu, C.-D. (2016). Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene, 36(9), 1223–1231. doi:10.1038/onc.2016.287
20. Beketova, E., Owens, J.L., Asberry, A.M. et al. (2021). PRMT5: a putative oncogene and therapeutic target in prostate cancer. Cancer Gene Ther. https://doi.org/10.1038/s41417-021-00327-3
21. Chiang, K., Zielinska, A. E., Shaaban, A. M., Sanchez-Bailon, M. P., Jarrold, J., Clarke, T. L., … Davies, C. C. (2017). PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Reports, 21(12), 3498–3513. doi:10.1016/j.celrep.2017.11.09
22. Bao X, Zhao S, Liu T, Liu Y, Liu Y, Yang X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J Histochem Cytochem. 2013; 61:206–17. https://doi. org/10.1369/0022155413475452.
23. Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007; 26:3558–69. https://doi.org/10.1038/sj.emboj.7601794.
24. Park JH, Szemes M, Vieira GC, Melegh Z, Malik S, Heesom KJ, Von Wallwitz-Freitas L, Greenhough A, Brown KW, Zheng YG, Catchpoole D, Deery MJ, Malik K. Protein arginine methyltransferase 5 is a key regulator of the MYCN oncoprotein in neuroblastoma cells. Mol Oncol. 2015; 9:617–27. https://doi.org/10.1016/j.molonc.2014.10.015.
25. Han X, Li R, Zhang W, Yang X, Wheeler CG, Friedman GK, Province P, Ding Q, You Z, Fathallah-Shaykh HM, Gillespie GY, Zhao X, King PH, Nabors LB. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J Neurooncol. 2014; 118:61–72. https://doi.org/10.1007/s11060-014-1419-0.
26. Sachamitr, P., Ho, J.C., Ciamponi, F.E. et al. PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat Commun 12, 979 (2021). https://doi.org/10.1038/s41467-021-21204-5
27. Nicholas C, Yang J, Peters SB, Bill MA, Baiocchi RA, Yan F, Sïf S, Tae S, Gaudio E, Wu X, Grever MR, Young GS, Lesinski GB. PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1.). PLoS One. 2013; 8:e74710. https://doi. org/10.1371/journal.pone.0074710.
28. Liu, X., He, J., Mao, L., Zhang, Y., Cui, W., Duan, S., … Huang, G. (2020). EPZ015666, a selective protein arginine methyltransferase 5 (PRMT5) inhibitor with an antitumour effect in retinoblastoma. Experimental Eye Research, 108286. doi:10.1016/j.exer.2020.108286
29. Marjon, K., Cameron, M. J., Quang, P., Clasquin, M. F., Mandley, E., Kunii, K., … Marks, K. M. (2016). MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Reports, 15(3), 574–587.doi:10.1016/j.celrep.2016.03.04
30. Kryukov, G. V., Wilson, F. H., Ruth, J. R., Paulk, J., Tsherniak, A., Marlow, S. E., Vazquez, F., Weir, B. A., Fitzgerald, M. E., Tanaka, M., Bielski, C. M., Scott, J. M., Dennis, C., Cowley, G. S., Boehm, J. S., Root, D. E., Golub, T. R., Clish, C. B., Bradner, J. E., Hahn, W. C., … Garraway, L. A. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science (New York, N.Y.), 351(6278), 1214–1218. https://doi.org/10.1126/science.aad5214
31. Mavrakis, K. J., McDonald, E. R., Schlabach, M. R., Billy, E., Hoffman, G. R., deWeck, A., … Sellers, W. R. (2016). Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science, 351(6278), 1208–1213.doi:10.1126/science.aad5944
32. Chan-Penebre, E., Kuplast, K. G., Majer, C. R., Boriack-Sjodin, P. A., Wigle, T. J., Johnston, L. D., … Duncan, K. W. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nature Chemical Biology, 11(6), 432–437. doi:10.1038/nchembio.1810
33. Kalev, P., Hyer, M. L., Gross, S., Konteatis, Z., Chen, C. C., Fletcher, M., Lein, M., Aguado-Fraile, E., Frank, V., Barnett, A., Mandley, E., Goldford, J., Chen, Y., Sellers, K., Hayes, S., Lizotte, K., Quang, P., Tuncay, Y., Clasquin, M., Peters, R., … Marjon, K. (2021). MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent mRNA Splicing and Inducing DNA Damage. Cancer cell, 39(2), 209–224.e11. https://doi.org/10.1016/j.ccell.2020.12.010
34. Bai, J., Gao, Y., Chen, L., Yin, Q., Lou, F., Wang, Z., … Wang, H. (2018). Identification of a natural inhibitor of methionine adenosyltransferase 2A regulating one-carbon metabolism in keratinocytes. EBioMedicine.doi:10.1016/j.ebiom.2018.12.036
35. Aronow, M. E., Topham, A. K., & Singh, A. D. (2018). Uveal Melanoma: 5-Year Update on Incidence, Treatment, and Survival (SEER 1973-2013). Ocular oncology and pathology, 4(3), 145–151. https://doi.org/10.1159/000480640
36. Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., de Tribolet, N., Weller, M., Kros, J. M., Hainfellner, J. A., Mason, W., Mariani, L., Bromberg, J. E., Hau, P., Mirimanoff, R. O., Cairncross, J. G., Janzer, R. C., & Stupp, R. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine, 352(10), 997–1003. https://doi.org/10.1056/NEJMoa043331
37. Wick, W., Platten, M., Meisner, C., Felsberg, J., Tabatabai, G., Simon, M., Nikkhah, G., Papsdorf, K., Steinbach, J. P., Sabel, M., Combs, S. E., Vesper, J., Braun, C., Meixensberger, J., Ketter, R., Mayer-Steinacker, R., Reifenberger, G., Weller, M., & NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society (2012). Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. The Lancet. Oncology, 13(7), 707–715. https://doi.org/10.1016/S1470-2045(12)70164-X
38. Reifenberger, G., Hentschel, B., Felsberg, J., Schackert, G., Simon, M., … Schnell, O. (2012). Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. International Journal of Cancer, 131(6), 1342–1350. doi:10.1002/ijc.27385
39. Fuentes-Fayos, A. C., Vázquez-Borrego, M. C., Jiménez-Vacas, J. M., Bejarano, L., Pedraza-Arévalo, S., L-López, F., Blanco-Acevedo, C., Sánchez-Sánchez, R., Reyes, O., Ventura, S., Solivera, J., Breunig, J. J., Blasco, M. A., Gahete, M. D., Castaño, J. P., & Luque, R. M. (2020). Splicing machinery dysregulation drives glioblastoma development/aggressiveness: oncogenic role of SRSF3. Brain : a journal of neurology, 143(11), 3273–3293. https://doi.org/10.1093/brain/awaa273
40. Jiménez-Vacas, J. M., Herrero-Aguayo, V., Montero-Hidalgo, A. J., Gómez-Gómez, E., Fuentes-Fayos, A. C., León-González, A. J., Sáez-Martínez, P., Alors-Pérez, E., Pedraza-Arévalo, S., González-Serrano, T., Reyes, O., Martínez-López, A., Sánchez-Sánchez, R., Ventura, S., Yubero-Serrano, E. M., Requena-Tapia, M. J., Castaño, J. P., Gahete, M. D., & Luque, R. M. (2020). Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. EBioMedicine, 51, 102547. https://doi.org/10.1016/j.ebiom.2019.11.008
41. Korfel, A., & Schlegel, U. (2013). Diagnosis and treatment of primary CNS lymphoma. Nature Reviews Neurology, 9(6), 317–327. doi:10.1038/nrneurol.2013.83
42. Dewson G, Kluck RM. (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 122(Pt 16):2801-2808.
43. Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM. (2008) To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell. 30(3):369-380.
44. Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM. (2012) Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 19(4):661-670.
45. Uren RT, O'Hely M, Iyer S, Bartolo R, Shi MX, Brouwer JM, Alsop AE, Dewson G, Kluck RM. (2017) Disordered clusters of Bak dimers rupture mitochondria during apoptosis. eLife. 6.
46. Hockings, C., Alsop, A. E., Fennell, S. C., Lee, E. F., Fairlie, W. D., Dewson, G., & Kluck, R. M. (2018). Mcl-1 and Bcl-xL sequestration of Bak confers differential resistance to BH3-only proteins. Cell Death & Differentiation.doi:10.1038/s41418-017-0010-6
47. Turenne GA, Paul P, Laflair L, Price BD. (2001) Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal serine residues of p53. Oncogene. 20(37):5100-5110.
48. Konopleva, M., Contractor, R., Tsao, T., Samudio, I., Ruvolo, P. P., Kitada, S., … Andreeff, M. (2006). Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell, 10(5), 375–388.doi:10.1016/j.ccr.2006.10.006
49. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L et al. (2016) Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 374(4):311-322.
50. Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, Leverson JD, Lam LT. (2017) Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 17(1):399.
51. Hanamura I. (2021). Gain/Amplification of Chromosome Arm 1q21 in Multiple Myeloma. Cancers, 13(2), 256. https://doi.org/10.3390/cancers13020256
52. Gupta, V. A., Barwick, B. G., Matulis, S. M., Shirasaki, R., Jaye, D. L., Keats, J. J., Oberlton, B., Joseph, N. S., Hofmeister, C. C., Heffner, L. T., Dhodapkar, M. V., Nooka, A. K., Lonial, S., Mitsiades, C. S., Kaufman, J. L., & Boise, L. H. (2021). Venetoclax sensitivity in multiple myeloma is associated with B-cell gene expression. Blood, 137(26), 3604–3615. https://doi.org/10.1182/blood.2020007899
53. Xiang, Z., Luo, H., Payton, J. E., Cain, J., Ley, T. J., Opferman, J. T., & Tomasson, M. H. (2010). Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. The Journal of clinical investigation, 120(6), 2109–2118. https://doi.org/10.1172/JCI39964
54. Ramsey, H. E., Fischer, M. A., Lee, T., Gorska, A. E., Arrate, M. P., Fuller, L., … Savona, M. R. (2018). A Novel MCL-1 Inhibitor Combined with Venetoclax Rescues Venetoclax Resistant Acute Myelogenous Leukemia. Cancer Discovery, CD–18–0140.doi:10.1158/2159-8290.cd-18-0140
55. Luedtke, D. A., Niu, X., Pan, Y., Zhao, J., Liu, S., Edwards, H., Chen, K., Lin, H., Taub, J. W., & Ge, Y. (2017). Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal transduction and targeted therapy, 2, 17012.
56. Caenepeel, S., Brown, S. P., Belmontes, B., Moody, G., Keegan, K. S., Chui, D., Whittington, D. A., Huang, X., Poppe, L., Cheng, A. C., Cardozo, M., Houze, J., Li, Y., Lucas, B., Paras, N. A., Wang, X., Taygerly, J. P., Vimolratana, M., Zancanella, M., Zhu, L., … Hughes, P. E. (2018). AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer discovery, 8(12), 1582–1597. https://doi.org/10.1158/2159-8290.CD-18-0387
57. Thomas, R. L., Roberts, D. J., Kubli, D. A., Lee, Y., Quinsay, M. N., Owens, J. B., Fischer, K. M., Sussman, M. A., Miyamoto, S., & Gustafsson, Å. B. (2013). Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes & development, 27(12), 1365–1377. https://doi.org/10.1101/gad.215871.113
58. Wang, X., Bathina, M., Lynch, J., Koss, B., Calabrese, C., Frase, S., Schuetz, J. D., Rehg, J. E., & Opferman, J. T. (2013). Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes & development, 27(12), 1351–1364. https://doi.org/10.1101/gad.215855.113
59. Brinkmann, K., Grabow, S., Hyland, C. D., Teh, C. E., Alexander, W. S., Herold, M. J., & Strasser, A. (2017). The combination of reduced MCL-1 and standard chemotherapeutics is tolerable in mice. Cell death and differentiation, 24(12), 2032–2043. https://doi.org/10.1038/cdd.2017.125
60. Spencer, A., Rosenberg, A. S., Jakubowiak, A., Raje, N., Chatterjee, M., Trudel, S., … Roberts, A. (2019). A Phase 1, First-in-Human Study of AMG 176, a Selective MCL-1 Inhibitor, in Patients With Relapsed or Refractory Multiple Myeloma. Clinical Lymphoma Myeloma and Leukemia, 19(10), e53–e54.doi:10.1016/j.clml.2019.09.081
61. Fernando, T. M., Piskol, R., Bainer, R., Sokol, E. S., Trabucco, S. E., Zhang, Q., … Yauch, R. L. (2020). Functional characterization of SMARCA4 variants identified by targeted exome-sequencing of 131,668 cancer patients. Nature Communications, 11(1). doi:10.1038/s41467-020-19402-8
62. Ho, A. S., Kannan, K., Roy, D. M., Morris, L. G. T., Ganly, I., Katabi, N., … Chan, T. A. (2013). The mutational landscape of adenoid cystic carcinoma. Nature Genetics, 45(7), 791–798.doi:10.1038/ng.2643
Have feedback? Tweet or DM me @blackseedbio on X, or contact me here.
If you liked this article and want to get notified when updates are available, consider subscribing (it's free, and you can unsubscribe at any time).
If you would like to donate to support the website, please click here.